Prostap 3 Dcs
4
Package Leaflet: Information for the user
Revision date: 10.10.2016 Leaflet reference:
PROST3 PIL
Prostap® 3 DCS 11.25 mg Powder and Solvent for Prolonged-release Suspension for Injection in Pre-filled Syringe
(leuprorelin acetate)
Your medicine is known by the above name, but will be referred to as Women only: Prostap® 3 throughout this leaflet.
Read all of this leaflet carefully before you start using this medicine because it contains important information for you.
• Keep this leaflet. You may need to read it again.
• If you have any further questions, ask your doctor or pharmacist.
• This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
• If you get any side effects talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section
4.
In this leaflet:
1. What Prostap® 3 is and what it is used for
2. What you need to know before you use Prostap® 3
3. How to take Prostap® 3
4. Possible side effects
5. How to store Prostap® 3
6. Contents of the pack and other information
1. WHAT PROSTAP® 3 IS AND WHAT IT IS USED FOR
Prostap® 3 is a synthetic hormone which can be used to reduce the levels of testosterone and estrogen circulating in the body.
Prostap® 3 is used to treat prostate cancer in men and endometriosis in women.
Use in children:
Prostap® 3 is a synthetic hormone which can be used to reduce the levels of testosterone and estrogen circulating in the body Prostap® 3 is used to treat premature puberty which is caused by a release of certain hormones from the pituitary gland (central precocious puberty) in girls under 9 years of age and boys under 10 years of age.
2. WHAT YOU NEED TO KNOW BEFORE YOU USE PROSTAP® 3
Use in children: Your doctor will make a precise diagnosis of central precocious puberty.
Do not take Prostap® 3:
• If you are allergic (hypersensitive) to leuprorelin acetate (Prostap® SR or Prostap® 3) or any of the other ingredients of Prostap® 3 (listed in section 6).
• If you are pregnant, planning to become pregnant or are breastfeeding.
• If you have abnormal vaginal bleeding which you have not discussed with your doctor.
• In girls with central precocious puberty
- if the girl to be treated is pregnant or breast-feeding.
- if the girl has undiagnosed vaginal bleeding.
Warnings and Precautions:
Both men and women:
• If you are diabetic Prostap® 3 can aggravate existing diabetes therefore diabetes patients may need more frequent monitoring of the blood glucose levels.
• If you have diabetes or suffer from heart problems, you should tell your doctor.
• If you are at an increased risk of thinning of the bones (osteoporosis) you should tell your doctor before taking Prostap® 3. Risk factors include:
• If you or any of your close family have thinning of the bones.
• If you drink excessive amounts of alcohol, and/or smoke heavily.
• If you take drugs for epilepsy or have taken steroids such as hydrocortisone or prednisolone for a long time.
• There have been reports of depression in patients taking Prostap® 3 which may be severe. If you are taking Prostap® 3 and develop depressed mood, inform your doctor.
• If you are a woman with submucous fibroids (benign tumours in the muscle underneath the lining of the womb), Prostap® 3 can cause severe bleeding when the fibroids break-down.
Contact your doctor immediately if you experience severe or unusual bleeding or pain.
• If you are a woman and continue to have periods (menstruate) after starting treatment with Prostap® 3 you should tell your doctor.
• If you are a woman of child-bearing age, you should use non hormonal contraception whilst receiving Prostap® 3. Although Prostap® 3 causes periods to stop, it is not itself a contraceptive. If you are unsure about this talk to your doctor.
Men only:
• In the rare event of an abscess occurring at the injection site your doctor may measure your testosterone levels as there could be reduced absorption of leuprorelin from the injection site.
• If you are a man with urinary obstruction or spinal cord compression. Your doctor will supervise you closely for the first few weeks of treatment.
• If you are a man with prostate cancer, and have had injections of a synthetic hormone in the past that has not worked, or you have had an operation to remove your testicles you should tell your doctor.
• Please tell your doctor if you have any of the following: Any heart or blood vessel conditions, including heart rhythm problems (arrhythmia), or are being treated with medicines for these conditions. The risk of heart rhythm problems may be increased when using Prostap® 3.
In children:
• In the event of a sterile abscess at the injection site (mostly reported after injection into the muscle) your doctor will monitor your hormone levels as there could be reduced absorption of leuprorelin from the injection site.
• If the child has progressive brain tumour your doctor will decide if treatment with leuprorelin is appropriate.
In girls with central precocious puberty:
• After the first injection vaginal bleeding (spotting) and discharge may occur as a sign of hormone withdrawal. Vaginal bleeding beyond the first/second month of treatment needs to be investigated.
• Bone density may decrease during treatment of central precocious puberty with Leuprorelin 3 Month Depot. However, after treatment is stopped, subsequent bone mass growth is preserved and peak bone mass in late adolescence does not seem to be affected by treatment.
• Often sterile abscesses at the injection site occurred when Leuprorelin 3 Month Depot is administered in higher dosages than recommended and when it is administered into the muscle. Your doctor will therefore administer the medicinal product under the skin of e.g. abdomen, bottom or thigh.
• Discontinuation of treatment may lead to a slipping of the growth plate of the thigh bone. A possible cause could be a weakness of the growth plate due to a lower concentration of female sexual hormones during treatment.
Other medicines and Prostap® 3
Please tell your doctor or pharmacist if you are taking or have recently taken any other medicines, including medicines obtained without a prescription.
Prostap® 3 might interfere with some medicines used to treat heart rhythm problems (e.g. quinidine, procainamide, amiodarone and sotalol) or might increase the risk of heart rhythm problems when used with some other drugs (e.g. methadone (used for pain relief and part of drug addiction detoxification), moxifloxacin (an antibiotic), antipsychotics used for serious mental illnesses).
Prostap® 3 with food and drink
Prostap® 3 can be taken with or without food.
Pregnancy and breastfeeding
Prostap® 3 must not be administered in pregnant or breast-feeding women or girls (see also section “Do not use Prostap® 3“).
Driving and using machines
Do not drive or operate machinery if you experience drowsiness, dizziness or visual disturbances whilst being treated with Prostap® 3.
3. HOW TO TAKE PROSTAP® 3
The doctor or nurse will give you an injection of Prostap® 3.
The injection will normally be given in your arm, thigh or abdomen. The injection site should be varied at regular intervals.
You will normally be given an injection once every 3 months.
If you have endometriosis you will be given an injection of Prostap® 3 for a period of 6 months only and treatment will be initiated during the first five days of the menstrual cycle.
Use in children
Treatment of children should be under the overall supervision of the paediatric endocrinologist.
The dosing scheme needs to be adapted individually.
The recommended starting dose is dependent on the body weight:
at Children with a body weight 20 kg or more
Unless prescribed otherwise, 1 ml Prostap® 3 (11,25 mg leuprorelin
acetate) is administered every 3 months under the skin of e.g. abdomen,
bottom or thigh as a single injection.
bt Children with a body weight less than 20 kg
Taking into account the clinical activity of the central precocious puberty in these rare cases, the following applies:
Unless prescribed otherwise, 0,5 ml Prostap® 3 (5,625 mg leuprorelin acetate) are administered every 3 months under the skin of e.g. abdomen, bottom or thigh as a single injection. The remainder of the suspension should be discarded. Your doctor will monitor the child’s weight gain.
Depending on the central precocious puberty activity, your doctor may increase the dosage in the presence of inadequate suppression (e.g. vaginal bleeding). Your doctor will determine the minimal effective dose with the help of a blood test.
The duration of treatment depends on the clinical signs at the start of treatment or during the course of treatment and is decided by your doctor together with the legal guardian and, if appropriate, the treated child. Your doctor will determine the bone age of the child in regular intervals.
In girls with bone maturation of older than 12 years and boys with bone maturation of older than 13 years your doctor will consider discontinuing the treatment, depending on the clinical effects in your child.
In girls, pregnancy should be excluded before the start of treatment. The occurrence of pregnancy during treatment cannot be generally excluded. In such cases, please talk to your doctor.
The therapy is a long-term treatment, adjusted individually. Please arrange with your doctor that Prostap® 3 is administered as precisely as possible in regular 3-monthly periods. An exceptional delay of the injection date for a few days (90 ± 2 days) does not influence the result of the therapy.
If you miss an injection
As soon as you realise you have missed an injection, contact your doctor who will be able to give you your next injection.
Women only:
If a Prostap® 3 injection is missed, breakthrough bleeding or ovulation may occur with the potential for conception. If you think you may be pregnant you should stop using Prostap® 3 and contact your doctor immediately.
4. POSSIBLE SIDE EFFECTS
Like all medicines, Prostap® 3 can cause side effects, although not everybody gets them.
Contact your doctor immediately or go to hospital:
• If you develop a severe rash, itching or shortness of breath or difficulty breathing. These could be symptoms of a severe allergic reaction.
Tell your doctor:
• If you get a severe headache which does not get better when you take painkillers.
• If you suffer from any unexplained bruising or bleeding or feel generally unwell whilst taking Prostap® 3. Although rare, these could be symptoms of changes in the number of red or white blood cells.
If any of the following side effects get serious, or if you notice any side effects not listed in this leaflet, speak to your doctor or pharmacist:
Men:
• When men with prostate cancer first start treatment with Prostap® 3, levels of testosterone can increase and in some people this may cause a temporary increase in local pain. In some cases, to prevent this from happening, your doctor may give you another type of drug such as cyproterone acetate or flutamide before and just after your first Prostap® 3 injection. If you do get worsening pain, weakness or loss of feeling in your legs or difficulty passing urine, contact your doctor immediately.
• If you have an existing pituitary lesion, there may be an increased risk of loss of blood to the area, which may cause permanent damage. This is very rare (may affect more than 1 in 10,000 people).
• Blood sugar levels may be altered during treatment with Prostap® 3, which may affect control in diabetic patients and require more frequent monitoring.
• If you have a blood test your doctor may notice a change in blood lipid (cholesterol) levels or in values for tests on how the liver is working. These changes do not usually cause any symptoms.
Very common (may affect more than 1 in 10 people)
Weight changes, hot flushes, sweating, muscle weakness, bone pain, loss of interest in sexual intercourse, inability to have an erection, a reduction in size and function of the testes, tiredness or skin reactions at the injection site (these include skin hardening, redness, pain, abscesses, swelling, nodules, ulcers and skin damage).
Common (may affect up to 1 in 10 people)
Loss of appetite, difficulty sleeping, depression, mood changes (with longterm use), headache, nausea, abnormalities in liver function or liver blood tests, joint pain, swelling of the breast tissue or swelling in your ankles.
Uncommon (may affect more than 1 in 100 people)
Mood changes (with short-term use), dizziness, tingling in the hands or feet, diarrhoea, vomiting, muscle ache or weakness in the legs.
Not known (frequency cannot be estimated from the available data)
Blood tests may show anaemia (low red cell counts), low counts in white cells or platelets, allergic reactions (may include symptoms of rash, itching, wheals or a serious allergic reaction which causes difficulty breathing or dizziness), changes in blood lipids (cholesterol) or blood sugar, paralysis, seizure, altered vision, pounding heartbeats, changes in ECG (QT prolongation), blood clots in lungs, high or low blood pressure, jaundice, fracture of the spine, thinning of bone, difficulty passing urine, fever or chills.
Women:
• Many of the side effects of Prostap® 3 are related to the decrease in oestrogen level. Oestrogen level returns to normal after treatment is stopped. Common side effects include hot flushes, mood swings, depression and vaginal dryness. As can happen naturally when women reach the menopause, Prostap® 3 can cause a small amount of bone thinning.
Vaginal bleeding may occur during treatment.
• If you have an existing pituitary lesion, there may be an increased risk of loss of blood to the area, which may cause permanent damage. This is very rare (may affect more than 1 in 10,000 people).
• Blood sugar levels may be altered during treatment with Prostap® 3, which may affect control in diabetic patients and require more frequent monitoring.
• If you have a blood test your doctor may notice a change in blood lipid (cholesterol) levels or in values for tests on how the liver is working. These changes do not usually cause any symptoms.
Very common (may affect more than 1 in 10 people)
Difficulty sleeping, headaches or hot flushes Common (may affect up to 1 in 10 people)
Weight changes, mood changes, depression, tingling in hands or feet, dizziness, nausea, joint pain, muscle weakness, breast tenderness, changes in breast size, vaginal dryness, swelling in ankles or skin reactions at the injection site (these include skin hardening, redness, pain, abscesses, swelling, nodules, ulcers and skin damage)
Uncommon (may affect more than 1 in 100 people)
Loss of appetite, changes in blood lipids (cholesterol), altered vision, pounding heartbeats, diarrhoea, vomiting, abnormalities in liver blood tests, hair loss, muscle aches, fever, chills or tiredness
Not known (frequency cannot be estimated from the available data)
Blood tests may show anaemia (low red cell counts), low counts in white cells or platelets, allergic reactions (may include symptoms of rash, itching, wheals or a serious allergic reaction causing difficulty breathing or dizziness), changes in blood sugar, paralysis, blood clots in the lungs, high or low blood pressure, jaundice, abnormalities in liver function, fracture of the spine, seizure, thinning of bone or vaginal bleeding.
Children
In the initial phase of treatment, a short-term rise in the sex hormone levels occurs, followed by a fall to values within the prepuberty range.
Due to this effect, side effects may occur particularly at the start of treatment.
Common (may affect up to 1 in 10 people):
• mood swings
• headache
• abdominal pain / abdominal cramps
• feeling sick/vomiting
• acne
• vaginal bleeding
• spotting
• discharge
• injection site reactions
Very rare (may affect less than 1 in 10,000 people):
• general allergic reactions (fever, rash, itching)
• serious allergic reaction which causes difficulty in breathing or dizziness
• As with other medicinal products of this class: if you have an existing pituitary lesion, there may be an increased risk of loss of blood to the area, which may cause permanent damage.
Not known (frequency cannot be estimated from the available data)
• Seizure Notes:
In general, if vaginal bleeding (spotting) occurs with continued treatment (after possible withdrawal bleeding in the first month of treatment), this may be a sign of potential underdosage. Please tell your doctor if vaginal bleeding occurs.
Reporting of side effects
If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. You can also report side effects directly via the Yellow Card Scheme at: www.mhra.aov.uk/vellowcard.
By reporting side effects, you can help provide more information on the safety of this medicine.
5. HOW TO STORE PROSTAP 3
Keep out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the packaging.
Do not store above 25°C.
Do not refrigerate or freeze.
Store in the original container in order to protect from light.
Once mixed with the Sterile Solvent, the suspension must be used immediately.
If the pack has been opened or damaged, return it to your pharmacist.
Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help to protect the environment.
6. CONTENTS OF THE PACK AND OTHER INFORMATION What Prostap® 3 contains:
• Each syringe contains 11.25 mg leuprorelin acetate. When reconstituted with the solvent, the solution contains 11,25mg/2ml of leuprorelin acetate.
• The excipients in Prostap® 3 are: Polylactic acid which controls the release of the active ingredient into the body, and mannitol.
• The Sterile Solvent contains carmellose sodium, mannitol (E421), polysorbate 80 and water for injections.
What Prostap® 3 looks like and contents of the pack:
Prostap® 3 is a prolonged release powder for use in an injection.
The Sterile Solvent is a clear liquid, which is mixed with the Prostap® 3 Powder before injection.
One dual chamber pre-filled syringe containing a sterile white powder in the front chamber and a clear sterile solvent in the rear chamber. The syringe contains a syringe plunger and a 23 gauge syringe needle fitted with a safety device. The pack contains 2 swabs soaked in alcohol.
Product Licence Holder and Manufacturer:
Manufactured by Takeda Pharmaceutical Company Ltd., Japan and Procured from the EU and repackaged by Product Licence Holder: Beachcourse Limited., 20 Alliance Court, Alliance Road,
London W3 ORB, UK.
PL 16378/0583 |POM|
Revision date: 10.10.2016 Leaflet reference: PROST3 PIL
Prostap® is a registered trademark of Takeda Pharmaceutical Company Limited.
HEALTH PROFESSIONALS’ USER LEAFLET
Prostap® 3 DCS 11.25 mg Powder and Solvent for Prolonged-release Suspension for Injection in Pre-filled Syringe
(leuprorelin acetate)
Revision date: 10.10.2016 Ref: PROST3 MIL-1
1 NAME OF THE MEDICINAL PRODUCT
Prostap® 3 DCS 11.25 mg Powder and Solvent for Prolonged-release Suspension for Injection in Pre-filled Syringe
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Prostap® 3 Powder: Each syringe contains 11.25 mg leuprorelin acetate. When reconstituted with the solvent, the solution contains 11.25mg/2ml of leuprorelin acetate.
Sterile Solvent: Each ml contains carmellose sodium 5 mg, mannitol (E421) 50 mg, polysorbate 80 1 mg, acetic acid, glacial up to 0.05 mg and water for injections.
When reconstituted with Sterile Solvent, the suspension contains 11.25 mg/ml leuprorelin acetate.
For the full list of excipients, see section 6.1
3 PHARMACEUTICAL FORM
Powder and solvent for suspension for injection in pre-filled syringe Powder: A sterile, lyophilised, white, odourless powder.
Solvent: A clear, odourless, slightly viscous, sterile solvent.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
(i) Metastatic prostate cancer.
(ii) Locally advanced prostate cancer, as an alternative to surgical castration.
(iii) As an adjuvant treatment to radiotherapy in patients with high-risk localised or locally advanced prostate cancer.
(iv) As an adjuvant treatment to radical prostatectomy in patients with locally advanced prostate cancer at high risk of disease progression.
(v) As neo-adjuvant treatment prior to radiotherapy in patients with high-risk localised or locally advanced prostate cancer.
(vi) Management of endometriosis, including pain relief and reduction of endometriotic lesions.
In children:
Treatment of central precocious puberty (girls under 9 years of age, boys under 10 years of age). (See Section 5.1)
4.2 Posology and method of administration Posology
Prostate Cancer: The usual recommended dose is 11.25 mg presented as a three month depot injection and administered as a single subcutaneous injection at intervals of three months. The majority of patients will respond to this dosage. Prostap® 3 therapy should not be discontinued when remission or improvement occurs. As with other drugs administered regularly by injection, the injection site should be varied periodically.
Response to Prostap® 3 therapy should be monitored by clinical parameters and by measuring prostate-specific antigen (PSA) serum levels. Clinical studies with leuprorelin acetate have shown that testosterone levels increased during the first 4 days of treatment in the majority of non-orchidectomised patients. They then decreased and reached castrate levels by 2-4 weeks. Once attained, castrate levels were maintained as long as drug therapy continued. If a patient's response appears to be sub-optimal, then it would be advisable to confirm that serum testosterone levels have reached or are remaining at castrate levels. Transient increases in acid phosphatase levels sometimes occur early in the treatment period but usually return to normal or near normal values by the 4th week of treatment.
In patients treated with GnRH analogues for prostate cancer, treatment is usually continued upon development of castrate-resistant prostate cancer. Reference should be made to relevant guidelines.
Endometriosis: The recommended dose is 11.25 mg administered as a single intramuscular injection every 3 months for a period of 6 months only. Treatment should be initiated during the first 5 days of the menstrual cycle. In women receiving GnRH analogues for the treatment of endometriosis, the addition of hormone replacement therapy (HRT - an estrogen and progestogen) has been shown to reduce bone mineral density loss and vasomotor symptoms. Therefore, if appropriate, HRT should be co-administered with Prostap® 3 taking into account the risks and benefits of each treatment.
Elderly: As for adults.
Paediatric population
The treatment of children with leuprorelin acetate should be under the overall supervision of the paediatric endocrinologist.
The dosing scheme needs to be adapted individually.
The recommended starting dose is dependent on the body weight.
Children with a body weight > 20 kg
1 ml (11.25 mg leuprorelin acetate) suspension of 130.0 mg sustained-release microcapsules in 1 ml vehicle solution are administered every 3 months as a single subcutaneous injection.
Children with a body weight < 20 kg
In these rare cases the following dosage should be administered according to the clinical activity of the central precocious puberty:
0.5 ml (5.625 mg leuprorelin acetate) suspension of 130.0 mg sustained-release microcapsules in 1 ml vehicle solution are administered every 3 months as a single subcutaneous injection.
The remainder of the suspension should be discarded. The child's weight gain should be monitored.
Depending on the activity of the central precocious puberty, it may be necessary to increase the dosage in the presence of inadequate suppression (clinical evidence e.g. spotting or inadequate gonadotropin suppression in the LHRH test). The minimal effective 3-monthly dose to be administered should then be determined by means of the LHRH test.
Sterile abscesses at the injection site often occurred when leuprorelin acetate was administered intramuscularly at higher than the recommended dosages. Therefore, in such cases, the medicinal product should be administered subcutaneously (see 4.4).
It is recommended to use the lowest volumes possible for injections in children in order to decrease the inconvenience which is associated with the intramuscular/subcutaneous injection.
The duration of treatment depends on the clinical parameters at the start of treatment or during the course of treatment (final height prognosis, growth velocity, bone age and/or bone age acceleration) and is decided by the treating paediatrician together with the legal guardian and, if appropriate, the treated child. The bone age should be monitored during treatment at 6-12 month intervals. In girls with bone maturation of older than 12 years and boys with bone maturation of older than 13 years discontinuation of treatment should be considered taking into account the clinical parameters.
In girls, pregnancy should be excluded before the start of treatment. The occurrence of pregnancy during treatment cannot be generally excluded. In such cases, medical advice should be sought.
Note:
The administration interval should be 90 ± 2 days in order to prevent the recurrence of precocious puberty symptoms.
1

1 .To prepare for injection,
screw the plunger
rod into the end stopper until
the stopper
begins to turn.
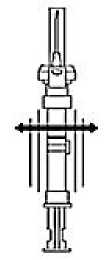

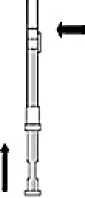
2. Remember to check if the needle is tight by twisting the needle cap clockwise.
Do not overtighten.
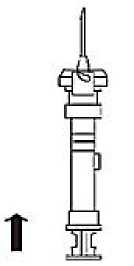
3. Holding the syringe upright, release the diluents by SLOWLY PUSHING the plunger until the middle stopper is at the blue line in the middle of the barrel.
NOTE: Pushing the plunger rod quickly or over the blue line will cause leakage of the suspension from the needle.
4. Gently tap the syringe on the palm keeping the syringe upright to thoroughly mix the particles to form a uniform suspension. The suspension will appear milky.
NOTE: Avoid hard tapping to prevent the generation of bubbles.
5. Remove the needle cap and advance the plunger to expel the air from the syringe.
|
UDSiltQ |
1 i n CUE* V p H H ii AFTER INJECTION |
|
Shin flaivS | |
|
6. At the time of injection, check the direction of the safety device (with round mark face up), as illustrated, and inject the entire contents of the syringe subcutaneously or intramuscularly as you would for a normal injection. |
7. Withdraw the needle from the patient. Immediately activate the safety device by pushing the arrow forward with the thumb or finger until the device is fully extended and a CLICK is heard or felt. |
Note: The suspension settles out very quickly following reconstitution and therefore the product should be mixed and used immediately.
4.3 Contraindications
Hypersensitivity to the active substance, any of the excipients listed in section 6.1 or to synthetic gonadotrophin releasing homone (Gn-RH) or Gn-RH derivatives.
Women: Prostap® 3 is contra-indicated in women who are or may become pregnant while receiving the drug. Prostap® 3 should not be used in women who are breastfeeding or have undiagnosed abnormal vaginal bleeding.
Men: There are no known contra-indications to the use of Prostap® 3 in men.
In girls with central precocious puberty:
- Pregnancy and lactation
- Undiagnosed vaginal bleeding
4.4 Special warnings and precautions for use
As would be expected with this class of drug, development or aggravation of diabetes may occur; therefore, diabetic patients may require more frequent monitoring of blood glucose during treatment with Prostap® 3. Epidemiological data have shown that during androgen deprivation therapy changes in the metabolic condition (e.g. reduction in glucose tolerance or aggravation of pre-existing diabetes) as well as an increased risk for cardiovascular diseases may occur. However, prospective data did not confirm the link between treatment with GnRH analogues and an increase in cardiovascular mortality. Patients at high risk for metabolic or cardiovascular diseases should be appropriately monitored.
Hepatic dysfunction and jaundice with elevated liver enzyme have been reported. Therefore, close observation should be made and appropriate measures taken if necessary.
Spinal fracture, paralysis and hypotension have been reported.
There is an increased risk of incident depression (which may be severe) in patients undergoing treatment with GnRH agonists, such as leuprorelin. Patients should be informed accordingly and treated as appropriate if symptoms occur. Postmarketing reports of seizures have been observed in patients treated with leuprorelin acetate and these events have been reported in both children and adults, and in those with or without a history of epilepsy, seizure disorders or risk disorders for seizures.
Men: In the initial stages of therapy, a transient rise in levels of testosterone, dihydrotestosterone and acid phosphatase may occur. In some cases, this may be associated with a “flare” or exacerbation of the tumour growth resulting in temporary deterioration of the patient's condition. These symptoms usually subside on continuation of therapy. “Flare” may manifest itself as systemic or neurological symptoms in some cases.
In order to reduce the risk of “flare”, an anti-androgen may be administered beginning 3 days prior to leuprorelin acetate therapy and continuing for the first two to three weeks of treatment. This has been reported to prevent the sequelae of an initial rise in serum testosterone.
In the rare event of an abscess occurring at the injection site, testosterone level should be monitored as there may be inadequate absorption of leuprorelin from the depot formulation.
Patients at risk of ureteric obstruction or spinal cord compression should be considered carefully and closely supervised in the first few weeks of treatment. These patients should be considered for prophylactic treatment with anti-androgens. Should urological/neurological complications occur, these should be treated by appropriate specific measures.
Long-term androgen deprivation either by bilateral orchiectomy or administration of GnRH analogues is associated with increased risk of bone loss which, in patients with additional risk factors, may lead to osteoporosis and increased risk of bone fracture. If an anti-androgen is used over a prolonged period, due attention should be paid to the contra-indications and precautions associated with its extended use.
Whilst the development of pituitary adenomas has been noted in chronic toxicity studies at high doses in some animal species, this has not been observed in long term clinical studies with leuprorelin acetate.
Androgen deprivation therapy may prolong the QT interval.
In patients with a history of or risk factors for QT prolongation and in patients receiving concomitant medicinal products that might prolong the QT interval (see section 4.5) physicians should assess the benefit risk ratio including the potential for Torsade de pointes prior to initiating Prostap® 3.
Women: During the early phase of endometriosis therapy, sex steroids temporarily rise above baseline because of the physiological effect of the drug. Therefore, a worsening of clinical signs and symptoms may be observed during the initial days of therapy, but these will dissipate with continued therapy.
When receiving GnRH analogues for the treatment of endometriosis, the addition of HRT (an estrogen and progestogen) has been shown to reduce bone mineral density loss and vasomotor symptoms (see ‘Posology and Method of Administration' section 4.2 for further information).
The induced hypo-estrogenic state results in a small loss in bone density over the course of treatment, some of which may not be reversible. The extent of bone demineralisation due to hypo-estrogenaemia is proportional to time and, consequently, is the adverse event responsible for limiting the duration of therapy to 6 months. The generally accepted level of bone loss with LHRH analogues such as Prostap® 3 is 5%. In clinical studies with Prostap® 3 the levels varied between 2.3% and 15.7% depending on the method of measurement. During one six-month treatment period, this bone loss should not be important. In patients with major risk factors for decreased bone mineral content such as chronic alcohol and/or tobacco use, strong family history of osteoporosis, or chronic use of drugs that can reduce bone mass such as anticonvulsants or corticosteroids, Prostap® 3 therapy may pose an additional risk. In these patients, the risks and benefits must be weighed carefully before therapy with Prostap® 3 is instituted.
In women with submucous fibroids there have been reports of severe bleeding following administration of Prostap® 3 as a consequence of the acute degeneration of the fibroids. Patients should be warned of the possibility of abnormal bleeding or pain in case earlier surgical intervention is required.
Prostap® 3 may cause an increase in uterine cervical resistance, which may result in difficulty in dilating the cervix for intrauterine surgical procedures.
Precautions
Men: Patients with urinary obstruction and patients with metastatic vertebral lesions should begin Prostap® 3 therapy under close supervision for the first few weeks of treatment.
Women: Since menstruation should stop with effective doses of Prostap® 3, the patient should notify her physician if regular menstruation persists.
In girls with central precocious puberty: Before starting the therapy, a precise diagnosis of idiopathic and/or neurogenic central precocious puberty is necessary.
The therapy is a long-term treatment, adjusted individually. Prostap® 3 should be administered as precisely as possible in regular 3-monthly periods. An exceptional delay of the injection date for a few days (90 ± 2 days) does not influence the results of the therapy.
In the event of a sterile abscess at the injection site (mostly reported after i.m. injection of higher than the recommended dosage) the absorption of leuprorelin acetate from the depot can be decreased. In this case the hormonal parameters (testosterone, oestradiol) should be monitored at 2-week intervals (see 4.2).
The treatment of children with progressive brain tumours should follow a careful individual appraisal of the risks and benefits.
The occurrence of vaginal bleeding, spotting and discharge after the first injection may occur as a sign of hormone withdrawal in girls. Vaginal bleeding beyond the first/second month of treatment needs to be investigated.
Bone mineral density (BMD) may decrease during GnRH therapy for central precocious puberty. However, after cessation of treatment subsequent bone mass accrual is preserved, and peak bone mass in late adolescence does not seem to be affected by treatment.
Slipped femoral epiphysis can be seen after withdrawal of GnRH treatment. The suggested theory is that the low concentrations of estrogen during treatment with GnRH agonists weakens the epiphysial plate. The increase in growth velocity after stopping the treatment subsequently results in a reduction of the shearing force needed for displacement of the epiphysis.
4.5 Interaction with other medicinal products and other forms of interaction
No interaction studies have been performed.
Since androgen deprivation treatment may prolong the QT interval, the concomitant use of Prostap® 3 with medicinal products known to prolong the QT interval or medicinal products able to induce Torsade de pointes such as class IA (e.g. quinidine, disopyramide) or class III (e.g. amiodarone, sotalol, dofetilide, ibutilide) antiarrhythmic medicinal products, methadone, moxifloxacin, antipsychotics, etc. should be carefully evaluated (see section 4.4).
4.6 Fertility, Pregnancy and lactation
Safe use of leuprorelin acetate in pregnancy has not been established clinically.
Studies in animals have shown reproductive toxicity (see section 5.3). Before starting treatment with Prostap® 3, pregnancy must be excluded. There have been reports of foetal malformation when Prostap® 3 has been given during pregnancy.
Prostap® 3 should not be used in women who are breastfeeding.
When used 3-monthly at the recommended dose, Prostap® 3 usually inhibits ovulation and stops menstruation. Contraception is not ensured, however, by taking Prostap® 3 and therefore patients should use non-hormonal methods of contraception during treatment.
Patients should be advised that if they miss successive doses of Prostap® 3, breakthrough bleeding or ovulation may occur with the potential for conception. Patients should be advised to see their physician if they believe they may be pregnant. If a patient becomes pregnant during treatment, the drug must be discontinued. The patient must be apprised of this evidence and the potential for an unknown risk to the foetus.
In girls with central precocious puberty: See section 4.3 Contraindications.
4.7 Effects on ability to drive and use machines
Prostap® 3 can influence the ability to drive and use machines due to visual disturbances and dizziness.
4.8 Undesirable effects
Adverse reactions seen with Prostap® 3 are due mainly to the specific pharmacological action, namely increases and decreases in certain hormone levels.
The following tables list adverse reactions with leuprorelin based on experience from clinical trials as well as from post-marketing experience. Adverse reactions are grouped by MedDRA System Organ Classes and frequency classification. Frequencies are defined as: very common (>1/10); common (>1/100 to <1/10); uncommon (>1/1,000 to <1/100); rare (>1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data).
Men: In cases where a “tumour flare” occurs after Prostap® 3 therapy, an exacerbation may occur in any symptoms or signs due to disease, for example, bone pain, urinary obstruction, weakness of the lower extremities and paraesthesia. These symptoms subside on continuation of therapy.
Tabulated list of adverse reactions
|
SOC |
Very common |
Common |
Uncommon |
Rare |
Very rare |
Not known |
|
Blood and lymphatic system disorders |
Anaemia (reported in medicinal products of this class), thrombo-cytopaenia, leucopenia | |||||
|
Immune system disorders |
hypersensitivity reactions (including rash, pruritus, urticaria and rarely, wheezing or interstitial pneumonitis, anaphylactic reactions) | |||||
|
Metabo lism and nutrition disorders |
weight fluctuation |
decreased appetite |
Lipids abnormal, glucose tolerance abnormal | |||
|
Psychia tric disorders |
insomnia, depression (see Section 4.4), mood changes (long-term use)** |
mood changes (short term use)** | ||||
|
Nervous system disorders |
headache (occasionaly severe) |
dizziness, parasthesiae |
pituitary apoplexy has been reported following initial administration in patients with pituitary adenoma |
paralysis (see Section 4.4), seizure | ||
|
Eye disorders |
visual impairment | |||||
|
Cardiac disorders |
palpitations, electrocardiogram QT prolonged (see Sections 4.4 and 4.5) | |||||
|
Vascular disorders |
hot flush |
pulmonary embolism, hypertension, hypotension (see Section 4.4) | ||||
|
Gastro intestinal disorders |
nausea |
diarrhoea, vomiting | ||||
|
Hepa tobiliary disorders |
hepatic function abnormal, liver function test abnormal (usually transient) |
jaundice | ||||
|
Skin and subcutan eous tissue disorders |
Hyperhy- drosis | |||||
|
Musculoskeletal, Connective tissue and bone disorders |
muscle weakness, bone pain |
arthralgia |
myalgia, weakness of lower extremities |
spinal fracture (see Section 4.4), reduction in bone mass which may occur with the use of GnRH agonists | ||
|
Renal and urinary disorders |
urinary tract obstruction | |||||
|
Repro ductive system and breast disorders |
Libido decree-sed, erectile dysfunction, testicular atrophy |
Gynaeco- mastia | ||||
|
General disorders and adminis tration site conditi ons |
Fatigue, injection site reaction, e.g., induration, erythema, pain, abscesses, swelling, nodules, ulcers and necrosis |
oedema peripheral |
pyrexia |
**mood changes (long term use: frequency of ‘common' and short term use: frequency of ‘uncommon')
SEE MIL-2 FOR THE REST OF INFORMATION
Women: Those adverse events occurring most frequently with Prostap® 3 are associated with hypo-estrogenism; the most frequently reported are hot flushes, mood swings including depression (occasionally severe), and vaginal dryness. Estrogen levels return to normal after treatment is discontinued.
The induced hypo-estrogenic state results in a small loss in bone density over the course of treatment, some of which may not be reversible (see Section 4.4).
Vaginal haemorrhage may occur during therapy due to acute degeneration of submucous fibroids (see Section 4.4).
Tabulated list of adverse reactions
Revision date: 10.10.2016 Ref: PROST3 MIL-2
In Children: In the initial phase of therapy, a short-term increase as flare-up of the sex hormone level occurs, followed by a decrease to values within the pre-pubertal range. Due to this pharmacological effect, adverse events may occur particularly at the beginning of treatment.
Tabulated list of adverse reactions
|
SOC |
Very common |
Common |
Uncommon |
Rare |
Very rare |
Not known |
|
Blood and lymphatic system disorders |
Anaemia (reported in medicinal products of this class), thrombo-cytopaenia, leucopenia | |||||
|
Immune system disorders |
hypersensitivity reactions (including rash, pruritus, urticaria and rarely, wheezing or interstitial pneumonitis, anaphylactic reactions) | |||||
|
Metabolis m and nutrition disorders |
weight fluctuation |
decreased appetite, lipids abnormal |
glucose tolerance abnormal, which may affect diabetic control | |||
|
Psychia tric disorders |
insomnia |
mood altered depression (see Section 4.4) | ||||
|
Nervous system disorders |
headache (occasiona ly severe) |
Parasthe- siae, dizziness |
pituitary haemorrha ge has been reported following initial administra tion in patients with pituitary adenoma |
paralysis (see Section 4.4), seizure | ||
|
Eye disorders |
visual impairment | |||||
|
Cardiac disorders |
palpitations | |||||
|
Vascular disorders |
hot flush |
pulmonary embolism, hypertension, hypotension (see Section 4.4) | ||||
|
Gastroint estinal disorders |
nausea |
diarrhoea, vomiting | ||||
|
Hepatobil iary disorders |
liver function test abnormal (usually transient) |
hepatic function abnormal, jaundice | ||||
|
Skin and Subcutaneous tissue disorders |
hair loss | |||||
|
Musculos keletal, Connective tissue and bone disorders |
arthralgia, muscle weakness |
myalgia |
spinal fracture (see Section 4.4), reduction in bone mass which may occur with the use of GnRH agonists | |||
|
Repro ductive system and breast disorders |
breast tenderness, breast atrophy, vulvovaginal dryness |
vaginal haemorrhage | ||||
|
General disorders and administr ation site conditi ons |
Oedema peripheral, injection site reaction e.g.injection site induration, erythema, pain, abscesses, swelling, nodules, ulcers and necrosis |
pyrexia, fatigue |
|
SOC |
Very common |
Common |
Uncommon |
Rare |
Very rare |
Not known |
|
Immune system disorders |
Hypersensitivity (fever, rash, e.g. itching, anaphylactic reactions) | |||||
|
Psychiatric disorders |
emotional lability | |||||
|
Nervous system disorders |
headache |
pituitary haemorrhage following initial administration in patients with pituitary adenoma |
seizure | |||
|
Gastrointestinal disorders |
abdominal pain / abdominal cramps, nausea/vomiting | |||||
|
Skin and subcutaneous tissue disorders |
acne | |||||
|
Reproductive system and breast disorders |
vaginal haemorrhage, spotting**, vaginal discharge | |||||
|
General disorders and administration site conditions |
injection site reactions |
** In general, the occurrence of vaginal spotting with continued treatment (subsequent to possible withdrawal bleeding in the first month of treatment) should be assessed as a sign of potential underdosage. The pituitary suppression should then be determined by an LHRH test.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard.
4.9 Overdose
No case of overdose has been reported.
In animal studies, doses of up to 500 times the recommended human dose resulted in dyspnoea, decreased activity and local irritation at the injection site. In cases of overdose, the patients should be monitored closely and management should be symptomatic and supportive.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Gonadotrophin-Releasing Hormone Analogues ATC code: L02AE 02
Prostap® 3 contains leuprorelin acetate, a synthetic nonapeptide analogue of naturally occurring gonadotrophin releasing hormone (GnRH) which possesses greater potency than the natural hormone. Leuprorelin acetate is a peptide and therefore unrelated to the steroids. Chronic administration results in an inhibition of gonadotrophin production and subsequent suppression of ovarian and testicular steroid secretion. This effect is reversible on discontinuation of therapy.
Administration of leuprorelin acetate results in an initial increase in circulating levels of gonadotrophins which leads to a transient increase in gonadal steroid levels in both men and women. Continued administration of leuprorelin acetate results in a decrease of gonadotrophin and sex steroid levels. In men serum testosterone levels, initially raised in response to early luteinising hormone (LH) release, fall to castrate levels in about 2-4 weeks.
Leuprorelin acetate is inactive when given orally.
A randomised, open-label, comparative multi-centre study was performed to compare the efficacy and safety of the 3.75 mg and 11.25 mg depots of leuprorelin acetate. 48% of patients included had locally advanced disease (T3N0M0), 52% of patients had metastatic disease.
Mean serum testosterone level fell below the threshold for chemical castration (0.5 ng/ml) at one month of treatment, continuing to decrease thereafter and stabilising at a value below the castration threshold. The decline in serum PSA mirrored that of serum testosterone in both groups.
In an open, prospective clinical trial involving 205 patients receiving 3.75 mg leuprorelin acetate on a monthly basis as treatment for metastatic prostate cancer, the long-term efficacy and safety of leuprorelin acetate was assessed. Testosterone levels were maintained below the castrate threshold over the 63-month follow up period. Median survival time exceeded 42.5 months for those receiving monotherapy and 30.9 months for those receiving leuprorelin acetate in combination with anti-androgens (this difference relating to baseline differences between groups).
In a meta-analysis involving primarily patients with metastatic disease, no statistically significant difference in survival was found for patients treated with LHRH analogues compared with patients treated with orchidectomy.
In another randomised, open-label, multi-centre comparative trial, leuprorelin acetate in combination with flutamide has been shown to significantly improve disease-free survival and overall survival when used as an adjuvant therapy to radiotherapy in 88 patients with highrisk localised (T1-T2 and PSA of at least 10 ng/mL or a Gleason score of at least 7), or locally advanced (T3-T4) prostate cancer. The optimum duration of adjuvant therapy has not been established. This US study used a higher dose of leuprorelin acetate (7.5 mg/month) which is therapeutically equivalent to the European licensed dose.
The use of a LHRH agonist may be considered after prostatectomy in selected patients considered at high risk of disease progression. There are no disease-free survival data or survival data with leuprorelin acetate in this setting.
Neoadjuvant leuprorelin acetate prior to radiotherapy has been shown to reduce prostate volume.
In children:
Reversible suppression of pituitary gonadotropin release occurs, with a subsequent decrease in oestradiol (E2) or testosterone levels to values in the pre-pubertal range.
Initial gonadal stimulation (flare-up) may cause vaginal bleeding in girls who are already post-menarchal at start of treatment. Withdrawal bleeding may occur at the start of treatment. The bleeding normally stops as treatment continues.
The following therapeutic effects can be demonstrated:
- Suppression of basal and stimulated gonadotropin levels to pre-pubertal levels;
- Suppression of prematurely increased sexual hormone levels to pre-pubertal levels and arrest of premature menstruation;
- Arrest/involution of somatic pubertal development (Tanner stages);
- Improvement/normalisation of the ratio of chronological age to bone age;
- Prevention of progressive bone age acceleration;
- Decrease of growth velocity and its normalization;
- Increase in final height.
Treatment result is the suppression of the pathologically, prematurely activated hypothalamic-pituitary-gonadal axis according to pre-pubertal age.
In a long-term clinical trial in children treated with leuprorelin at doses up to 15mg monthly for > 4 years resumption of pubertal progression were observed after cessation of treatment. Follow up of 20 female subjects to adulthood showed normal menstrual cycles in 80% and 12 pregnancies in 7 of the 20 subjects including multiple pregnancies for 4 subjects.
5.2 Pharmacokinetic properties
Leuprorelin acetate is well absorbed after subcutaneous and intramuscular injections. It binds to the LHRH receptors and is rapidly degraded. An initially high plasma level of leuprorelin acetate peaks at around 3 hours after a Prostap® 3 subcutaneous injection, followed by a decrease to maintenance levels in 7 to 14 days. Prostap® 3 provides continuous plasma levels for up to 117 days resulting in suppression of testosterone to below castration level within 4 weeks of the first injection in the majority of patients.
The metabolism, distribution and excretion of leuprorelin acetate in humans have not been fully determined.
In children:
Figure 1 presents the leuprorelin serum levels in children during the first 6 months of treatment following s.c. administration of leuprorelin acetate 3-month depot (two injections).
From the first injection, the leuprorelin serum levels increase reaching maximal serum levels at month 4 (294.79 pg/ml ± 105.42) and slightly decrease until month 6 (229.02 pg/ml ± 103.33).
295
275 --
2E-E- -
23E--
21E
175-
EE
13E
75-
EE
1 Mon:h
2 r.1on:hs
3 r.lGn:hs
4 r.1on:hs
5 r.1on:hs
6 r.1on:hs
Figure 1: Leuprorelin serum levels during the first six months of treatment with the leuprorelin acetate 3-month depot formulation (two s.c. injections) (n=42-43)
5.3 Preclinical safety data
A teratogenic effect has been observed in rabbits but not in rats.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
The other ingredients are:
Powder- Polylactic acid, mannitol
Sterile solvent- Carmellose sodium, mannitol, polysorbate 80 and water for injections.
6.2 Incompatibilities
Not applicable.
6.3 Shelf life
3 years unopened.
Once reconstituted with sterile solvent, the suspension should be administered immediately.
6.4 Special precautions for storage
Do not store above 25oC.
Do not refrigerate or freeze.
Store in the original container in order to protect from light.
6.5 Nature and contents of container
One dual chamber pre-filled syringe containing a sterile white powder in the front chamber and a clear sterile solvent in the rear chamber. The syringe contains a syringe plunger and a 23 gauge syringe needle fitted with a safety device. The pack contains 2 swabs soaked in alcohol.
6.6 Special precautions for disposal and other handling
Any unused product or waste material should be disposed of in accordance with local requirements.
Product Licence Holder and Manufacturer:
Manufactured by Takeda Pharmaceutical Company Ltd., Japan and Procured from the EU and repackaged by Product Licence Holder: Beachcourse Limited., 20 Alliance Court, Alliance Road, London W3 0RB, UK.
PL 16378/0583 |POM|
Revision date: 10.10.2016 Leaflet reference: PROST3 MIL-2
Prostap® is a registered trademark of Takeda Pharmaceutical Company Limited.
4