Feiba 500 U Powder And Solvent For Solution For Infusion
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
FEIBA 500 U powder and solvent for solution for infusion
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
FEIBA is presented as powder and solvent to prepare a solution for infusion containing 200600 mg human plasma protein with a Factor Eight Inhibitor Bypassing Activity of 500 U* per vial.
The final solution has an activity of approximately 25 U/ml when reconstituted with 20 ml of Sterilised Water for Injections.
FEIBA contains factors II, IX and X mainly in non-activated form as well as activated factor VII; factor VIII coagulant antigen (FVIII C:Ag) is present in a concentration of up to 0.1 U/l U FEIBA. The factors of the kallikrein-kinin system are present only in trace amounts, if at all.
*A solution containing 1 unit of FEIBA shortens the activated partial thromboplastin time (aPTT) of a factor VIII inhibitor plasma to 50% of the buffer value (blank).
For excipients, see 6.1.
3 PHARMACEUTICAL FORM
Powder and solvent for solution for infusion.
The product is presented as freeze-dried powder or friable solid of white to off-white or pale green colour.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
• Treatment of spontaneous bleeding and cover of surgical interventions in haemophilia A patients with Factor VIII inhibitors
• Treatment of spontaneous bleeding and cover of surgical interventions in non haemophiliacs with acquired inhibitors to Factor VIII
• Prophylaxis in haemophilia A patients with high-responding inhibitors and frequent joint bleeding
4.2 Posology and method of administration
Treatment should be initiated and supervised by a physician experienced in the management of haemophilia.
Posology
The dosage and duration of the therapy is dependent upon the severity of the disorder, the location and extent of the bleeding and the patient’s clinical condition.
Dosage and frequency of administration should always be guided by the clinical efficacy in each individual case.
As a general guide a dose of 50 to 100 U of FEIBA per kg bodyweight (bw) is recommended. A single dose of 100 U/kg body weight and a daily dose of 200 U/kg body weight should not be exceeded unless the severity of bleeding warrants and justifies the use of higher doses. See section 4.4.
The following table can be used to guide dosing in bleeding episodes and surgery.
|
Therapeutic indication |
Dose (U/kg/bw) |
Frequency of doses (hours) |
|
Spontaneous Bleeding | ||
|
Joint muscle and soft tissue haemorrhage Minor to moderate bleeding |
50-75 U/kg/bw |
Repeat every 12 hours. Treatment should be continued until clear signs of clinical improvement, such as relief of pain, reduction of swelling or mobilisation of the joint. |
|
Joint muscle and soft tissue haemorrhage Major bleeding |
100 U/kg/bw |
Repeat every 12 hours. Treatment should be continued until clear signs of clinical improvement, such as relief of pain, reduction of swelling or mobilisation of the joint. |
|
Mucous membrane bleeding |
50 U/kg/bw if haemorrhage does not stop, the dose may be increased to 100 U/kg/bw |
Repeat every 6 hours with careful monitoring of the patient (visible bleeding site, repeated measurements of haematocrit). |
|
Other severe haemorrhage (e.g. CNS) |
100 U/kg/bw |
Repeat every 12 hours. In individual cases FEIBA may be given at intervals of 6 hours until clear clinical improvement is achieved. |
|
Surgery | ||
|
Surgery |
50-100 U/kg/bw |
Repeat at intervals of up to 6 hours, then every 8-12 hours until wound healing. |
Bleeding prophylaxis
Limited experience has been published on the use of FEIBA in haemophilia A patients with high-responding inhibitors before and during immune tolerance induction (ITI) therapy, or after ITI failure.
For prevention of bleeding episodes during prophylaxis, dose 70 to 100 units per kg body weight every other day. Adjust dose based on the patient’s clinical response.
Paediatric population
The experience in children under 6 years of age is limited; the same dose regimen as in adults should be adapted to the child’s clinical condition.
Monitoring
• In case of inadequate response to treatment with the product, it is recommended that a platelet count be performed because a sufficient number of functionally intact platelets are considered to be necessary for the efficacy of the product.
• Due to the complex mechanism of action, no direct monitoring of active ingredients is available. Coagulation tests such as whole blood clotting time (WBCT) and the aPTT may not correlate with clinical improvement.
• Global hemostatic tests such as thromboelastogram (TEG) or thrombin generation assay (TGA) may be useful tools to monitor and optimize the treatment.
Method of Administration
Reconstitute the product for administration as described in section 6.6.
FEIBA must be administered as an intravenous injection or infusion. The rate of administration should ensure the comfort of the patient and should not exceed a maximum of 2 U/kg body weight per minute.
4.3 Contraindications
FEIBA must not be used in the following situations if therapeutic alternatives to FEIBA are available:
• Hypersensitivity to the product
• Disseminated intravascular coagulation (DIC)
• Acute thrombosis or embolism (including myocardial infarction)
See section 4.4.
4.4 Special warnings and precautions for use
WARNINGS
Risk of Thromboembolic Events
• Thromboembolic events, including disseminated intravascular coagulation (DIC), venous thrombosis, pulmonary embolism, myocardial infarction, and stroke, have occurred in the course of treatment with FEIBA.
Some of these events occurred with doses above 200 U/kg/day or in patients with other risk factors (including DIC, advanced atherosclerotic disease, crush injury or septicemia) for thromboembolic events. Concomitant treatment with recombinant Factor VIIa may increase the risk of developing a thromboembolic event. The possible presence of such risk factors should always be considered in patients with congenital and acquired hemophilia.
FEIBA should be used with particular caution in patients at risk of DIC, arterial or venous thrombosis. See Section 4.3.
At the first signs or symptoms of thromboembolic events, the infusion should be stopped immediately and appropriate diagnostic and therapeutic measures initiated.
A single dose of 100 U/kg body weight and a daily dose of 200 U/kg body weight should not be exceeded unless the severity of bleeding warrants and justifies the use of higher doses.
When used to stop bleeding, the product should be given only for as long as absolutely necessary to achieve the therapeutic goal.
Allergic-Type Hypersensitivity Reactions
• FEIBA can precipitate allergic-type hypersensitivity reactions that have included, urticaria, angioedema, gastrointestinal manifestations, bronchospasm, and hypotension; these reactions can be severe and can be systemic (e.g., anaphylaxis with urticaria and angioedema, bronchospasm, and circulatory shock). Other infusion reactions, such as chills, pyrexia, and hypertension have also been reported.
At the first sign or symptom of an infusion/hypersensitivity reaction, FEIBA administration should be stopped and medical care initiated as appropriate.
When considering reexposure to FEIBA in patients with known or suspected hypersensitivity to the product, the expected benefit and the risk of reexposure must be carefully weighed, taking into account the known or suspected type of the
patient’s hypersensitivity (allergic or nonallergic), including potential remedial and/or preventative therapy or alternative therapeutic agents.
See section 4.8.
Viral Safety
Standard measures to prevent infections resulting from the use of medicinal products prepared from human blood or plasma include selection of donors, screening of individual donations and plasma pools for specific markers of infection and the inclusion of effective manufacturing steps for the inactivation/removal of viruses. Despite this, when medicinal products prepared from human blood or plasma are administered, the possibility of transmitting infective agents cannot be totally excluded. This also applies to unknown or emerging viruses and other pathogens.
The measures taken are considered effective for enveloped viruses such as HIV, HBV, and HCV and for the nonenveloped virus HAV. The measures taken may be of limited value against nonenveloped viruses such as parvovirus B19. Parvovirus B19 infection may be serious for pregnant women (foetal infection) and for individuals with immunodeficiency or increased erythropoiesis (e.g. haemolytic anaemia).
Appropriate vaccination (against hepatitis A and B) should be considered for patients in regular/repeated receipt of plasma-derived products including FEIBA.
PRECAUTIONS
• Due to patient-specific factors the response to a bypassing agent can vary, and in a given bleeding situation patients experiencing insufficient response to one agent may respond to another agent. In case of insufficient response to one bypassing agent, use of another agent should be considered.
• Administration of FEIBA to patients with inhibitors may result in an initial “anamnestic” rise in inhibitor levels. Upon continued administration of FEIBA, inhibitors may decrease over time.
• After administration of high doses of FEIBA, the transitory rise of passively transferred Hepatitis B surface antibodies may result in misleading interpretation of positive results in serological testing.
• The amount of sodium in the maximum daily dose may exceed the recommended daily allowance of dietary sodium for patients on a low sodium diet. In these patients, the amount of sodium from the product should be calculated and taken into account when determining dietary sodium intake.
• FEIBA 500U/1000U contains approximately 80 mg sodium (calculated) per vial
• The recording of the product name and batch number is strongly recommended following each administration of this product in order to be able to identify the batch of product received.
Paediatrics
• Case reports and limited clinical trial data suggest that FEIBA can be used in children younger than 6 years of age.
4.5 Interaction with other medicinal products and other forms of interaction
No adequate and well-controlled studies of the combined or sequential use of FEIBA and recombinant Factor VIIa or antifibrinolytics have been conducted.
The possibility of thromboembolic events should be considered when systemic antifibrinolytics such as tranexamic acid and aminocaproic acid are used during treatment with FEIBA. Therefore, antifibrinolytics and FEIBA should be administered at least 6 hours apart.
In cases of concomitant rFVIIa use, according to available in vitro data and clinical observations a potential drug interaction may occur (potentially resulting in adverse events such as a thromboembolic event).
4.6 Fertility, pregnancy and lactation
There are no adequate data from the use of FEIBA in pregnant or lactating women.
Healthcare providers should balance the potential risks and only prescribe FEIBA if clearly needed, taking into consideration that pregnancy and the postpartum period confer an increased risk of thrombotic events, and several complications of pregnancy that are associated with an increased risk of DIC. Careful medical monitoring is required.
No animal reproduction studies have been conducted with FEIBA.
The effects of FEIBA on fertility have not been established in controlled clinical trials.
See section 4.4 for information on parvovirus B19 infection.
4.7 Effects on ability to drive and use machines
No effects on the ability to drive and use machines have been observed.
4.8 Undesirable effects
Adverse Reactions From Clinical Trials
The adverse reactions presented in this section have been reported from 2 studies with FEIBA for the treatment of bleeding episodes in pediatric and adult patients with hemophilia A or B and inhibitors to factors VIII or IX. One study also enrolled acquired hemophilia patients with factor VIII inhibitors (2 of 49 patients).
The adverse reactions presented in the table were reported in the original FEIBA studies (Hilgartner 1983, 2003; Sjamsoedin LJ. et al., 1981) for the treatment of bleeding episodes in hemophilia A or B patients with inhibitors to Factors VIII or IX and the randomized, prospective prophylaxis study (090701) comparing prophylaxis with on-demand treatment.
|
System Organ Class (SOC) |
Preferred MedDRA |
Frequency |
|
(version 18.0) Term |
Category* | |
|
BLOOD AND LYMPHATIC |
Increase of inhibitor titer |
Unknown |
|
SYSTEM DISORDERS |
(anamnestic response)* a | |
|
IMMUNE SYSTEM |
Hypersensitivity |
Common |
|
DISORDERS | ||
|
NERVOUS SYSTEM |
Somnolence |
Unknown |
|
DISORDERS |
Dizziness |
Common |
|
Dysgeusia* |
Unknown | |
|
Headache |
Common | |
|
VASCULAR DISORDERS |
Hypotension |
Common |
|
RESPIRATORY, THORACIC, |
Dyspnea* |
Unknown |
|
AND MEDIASTINAL | ||
|
DISORDERS | ||
|
GASTROINTESTINAL |
Nausea* |
Unknown |
|
DISORDERS | ||
|
SKIN AND SUBCUTANEOUS |
Rash |
Common |
|
GENERAL DISORDERS AND |
Chills* |
Unknown |
|
ADMINISTRATION SITE |
• * Pyrexia |
Unknown |
|
CONDITIONS |
Chest pain* |
Unknown |
|
Chest discomfort* |
Unknown | |
|
INVESTIGATIONS |
Hepatitis B surface |
Common |
|
antibody positive |
Legend: ADR frequency is based upon the following scale: Very Common (>1/10); Common (>1/100 -<1/10), Uncommon (>1/1,000 - <1/100), Rare (>1/10,000 - <1/1,000), Very Rare (<1/10,000)
*A precise estimate of the rate of these adverse reactions is not possible from the available data. a Increase of inhibitor titer (anamnestic response) [not a MedDRA PT] is the rise of previously existing inhibitor titers occurring after the administration of FEIBA. See Section 4.4.
Post-marketing Adverse Reactions
The following adverse reactions have been reported in the post-marketing experience, listed by MedDRA (version 18.0) System Organ Class (SOC), then by Preferred Term in order of severity, where feasible.
BLOOD AND LYMPHATIC SYSTEM DISORDERS: Disseminated intravascular coagulation
IMMUNE SYSTEM DISORDERS: Anaphylactic reaction
NERVOUS SYSTEM DISORDERS: Paresthesia, Thrombotic stroke, Embolic stroke
CARDIAC DISORDERS: Myocardial infarction, Tachycardia
VASCULAR DISORDERS: Thrombosis, Venous thrombosis, Arterial thrombosis, Hypertension, Flushing
RESPIRATORY, THORACIC, AND MEDIASTINAL DISORDERS: Pulmonary embolism, Bronchospasm, Wheezing, Cough
GASTROINTESTINAL DISORDERS: Vomiting, Diarrhea, Abdominal discomfort
SKIN AND SUBCUTANEOUS TISSUE DISORDERS: Angioedema, Urticaria, Pruritus
GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS: Malaise, Feeling hot, Injection site pain
Class Reactions
Other symptoms of hypersensitivity reactions to plasma-derived products include lethargy and restlessness.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme at: www.mhra.gov.uk/yellowcard.
4.9 Overdose
Some of the reported thromboembolic events have occurred with doses above 200 U/kg. See section 4.4. If signs or symptoms of thromboembolic events are observed, the infusion should be stopped immediately and appropriate diagnostic and therapeutic measures initiated. See section 4.4.
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: activated prothrombin complex against factor VIII antibodies, ATC Code: B02B D03
Although FEIBA was developed in the early 1970s and its factor VIII inhibitor bypassing activity has been demonstrated both in vitro and in vivo, its active principle is still the subject of scientific debate. However, recent scientific work indicates a role of specific components of the activated prothrombin complex, zymogen prothrombin (F II) and activated Factor X (FXa), in the FEIBA mode of action.
Administration of FEIBA to patients with inhibitors may result in an initial “anamnestic” rise in inhibitor levels. Upon continued administration of FEIBA, inhibitors may decrease over time.
Clinical and published data suggest that the efficacy of FEIBA is not reduced.
5.2 Pharmacokinetic properties
Since FEIBA is composed of different coagulation factors, with varying half-lives for the single components, it is not possible to make any definite statement with regard to the pharmacokinetic properties of FEIBA.
5.3 Preclinical safety data
Based on the acute toxicity studies in factor VIII knockout mice and in normal mice and rats with doses exceeding the maximum daily dose in humans (i.e. >200 U/kg bw), it can be concluded that adverse effects related to FEIBA are primarily the result of hypercoagulation induced by the pharmacological properties of the product.
Repeated dose toxicity testing in animals is impracticable due to interference with developing antibodies to heterologous protein.
Since human plasma proteins are not seen to cause tumorigenic or mutagenic effects, experimental studies particularly in heterologous species are not considered necessary.
6 PHARMACEUTICAL PARTICULARS
6.1
List of excipients
Powder
Sodium Chloride Sodium Citrate Protein
Solvent
Sterilised Water for Injections
6.2 Incompatibilities
No compatibility studies have been performed with the product. Therefore, FEIBA must not be mixed with other medicinal products or solvents.
Coagulation factors derived from human plasma may be adsorbed by the inner surfaces of certain types of injection/infusion devices. If this were to occur, it could result in failure of therapy.
6.3 Shelf life
2 years. The reconstituted solution should be used immediately.
6.4 Special precautions for storage
Do not store above 25°C. Do not freeze.
Keep container in the outer carton in order to protect from light.
6.5 Nature and contents of container
FEIBA powder and solvent are supplied in vials (hydrolytic Type II surface treated soda lime glass) closed with halogenobutyl rubber stoppers and protective caps.
Each pack contains 1 vial each of FEIBA powder and sterile water, and a device for reconstitution (BAXJECT II Hi-Flow).
6.6
Special precautions for disposal
- Aseptic conditions are required during preparation of the FEIBA solution and administration.
-The BAXJECT II Hi-Flow is used to reconstitute the powder with the sterile water.
- To prepare the FEIBA solution, use only the sterile water and the reconstitution device provided in the pack. For administration the use of a luer lock syringe is recommended.
- FEIBA should be reconstituted just prior to administration. The solution should then be used immediately as the preparation contains no preservatives.
- After reconstitution, the solution should be inspected for particulate matter and discolouration prior to administration. Do not use solutions that are cloudy or have deposits.
- Do not use if the BAXJECT II Hi-Flow device, its sterile barrier system or its packaging is damaged or shows any sign of deterioration.
- Any unused solution or waste material should be disposed of in accordance with local requirements.
Reconstitution of powder: use aseptic technique as described below
1. Warm the FEIBA and solvent (sterilised water for injections) vials to room temperature (15 °C - 25 °C) if necessary.
2. Remove the protective caps from the FEIBA and sterile water and cleanse the rubber stoppers of both with alcohol swabs. Place the vials on a flat surface.
3. Open the BAXJECT II Hi-Flow device package by peeling away the paper lid without touching the inside (Fig a). Do not remove the device from the package.
4. Turn the package over and insert the clear plastic spike through the solvent stopper (Fig. b). Grip the package at its edge and pull the package off BAXJECT II Hi-Flow (Fig. c). Do not remove the blue cap from BAXJECT II Hi-Flow device.
5. With BAXJECT II Hi-Flow attached to the sterile water vial, invert the system so that the sterile water vial is on top of the device. Insert the purple plastic spike through the FEIBA stopper. The vacuum will draw the sterile water into the FEIBA vial (Fig d).
6. Swirl, but do not shake, the entire system gently until all material is dissolved. Ensure that FEIBA is completely dissolved, otherwise active material will not pass through the device filter.
Figure a

Figure b
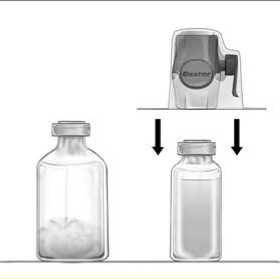
Figure c

Instructions for Injection/Infusion:
1. Remove the blue cap from BAXJECT II Hi-Flow. Take the syringe and tightly connect it to BAXJECT II Hi-Flow (DO NOT DRAW AIR INTO THE SYRINGE) (Fig. e). In order to ensure a tight connection between the syringe and BAXJECT II Hi-Flow, the use of a luer lock syringe is highly recommended (turn the syringe in a clockwise direction until the stop position when mounting).
2. Invert the system so that the dissolved product is on top. Draw the FEIBA solution into the syringe by pulling the plunger back SLOWLY and ensure that the tight connection between BAXJECT II Hi-Flow and the syringe is maintained throughout the whole pulling process (Fig. f).
3. Disconnect the syringe.
4. If foaming of the product in the syringe occurs, wait until the foam has collapsed. Slowly administer the solution intravenously with a winged set for injection (or a disposable needle).
Figure d

Figure e
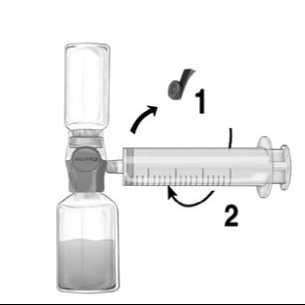
Figure f
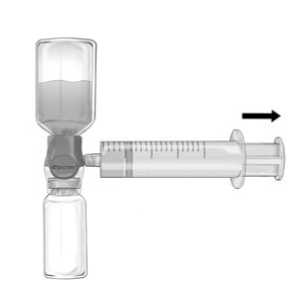
If devices other than those supplied with FEIBA are used, ensure use of an adequate filter.
Do not exceed an injection speed of 2 U FEIBA/kg body weight per minute.
7 MARKETING AUTHORISATION HOLDER
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vienna Austria
8 MARKETING AUTHORISATION NUMBER(S)
PL 34078/0002
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
17 October 1985
10 DATE OF REVISION OF THE TEXT
06/01/2016