Haemoctin 1000 Powder And Solvent For Solution For Injection
SUMMARY OF PRODUCT CHARACTERISTICS 1 NAME OF THE MEDICINAL PRODUCT
Haemoctin 1000
Powder and solvent for solution for injection
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Human plasma derived coagulation factor VIII
One vial contains nominally 250, 500 or 1000 IU human plasma derived coagulation factor VIII. Haemoctin 250 or Haemoctin 500 contains approximately 50 IU/ml human coagulation factor VIII when reconstituted with 5 or 10 ml of water for injections. Haemoctin 1000 contains approximately 100 IU/ml human coagulation factor VIII when reconstituted with 10 ml of water for injections.
The potency (IU) is determined using the European Pharmacopoeia chromogenic factor VIII coagulation assay. The specific activity of Haemoctin is approximately 100 IU/mg protein.
Produced from the plasma of human donors.
Excipient with known effect:
One vial contains up to 32.2 mg sodium (1.4 mmol).
For the full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Powder and solvent for solution for injection.
White powder and clear, colourless solvent for solution for injection.
CLINICAL PARTICULARS
4
4.1 Therapeutic indications
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII deficiency).
This preparation does not contain von Willebrand factor in pharmacologically effective quantities and is therefore not indicated in von Willebrand's disease.
4.2 Posology and method of administration
Treatment should be under the supervision of a physician experienced in the treatment of haemophilia.
Previously untreated patients No data are available.
Treatment monitoring
During the course of treatment, appropriate determination of factor VIII levels is advised to guide the dose to be administered and the frequency of repeated infusions. Individual patients may vary in their response to factor VIII, demonstrating different half-lives and recoveries. Dose based on bodyweight may require adjustment in underweight or overweight patients. In the case of major surgical interventions in particular, precise monitoring of the substitution therapy by means of coagulation analysis (plasma factor VIII activity) is indispensable.
When using an in vitro thromboplastin time (aPTT)-based one stage clotting assay for determining factor VIII activity in patients’ blood samples, plasma factor VIII activity results can be significantly affected by both the type of aPTT reagent and the reference standard used in the assay. Also there can be significant discrepancies between assay results obtained by aPTT-based one stage clotting assay and the chromogenic assay according to Ph. Eur. This is of importance particularly when changing the laboratory and/or reagents used in the assay.
Posology
The dose and duration of the substitution therapy depend on the severity of the factor VIII deficiency, on the location and extent of the bleeding and on the patient's clinical condition.
The number of units of factor VIII administered is expressed in International Units (IU), which are related to the current WHO concentrate standard for factor VIII products. Factor VIII activity in plasma
is expressed either as a percentage (relative to normal human plasma) or preferably in International Units (relative to an International Standard for factor VIII in plasma).
One International Unit (IU) of factor VIII activity is equivalent to that quantity of factor VIII in one ml of normal human plasma.
On demand treatment
The calculation of the required dose of factor VIII is based on the empirical finding that 1 International Unit (IU) factor VIII per kg body weight raises the plasma factor VIII activity by 1 % to 2 % of normal activity.
The required dose is determined using the following formula:
Required units = body weight (kg) x desired factor VIII rise (%) x 0.5
The amount to be administered and the frequency of administration should always be oriented to the clinical effectiveness in the individual case.
In the case of the following haemorrhagic events, the factor VIII activity should not fall below the given plasma activity level (in % of normal) in the corresponding period. The following table can be used to guide dosing in bleeding episodes and surgery:
|
Degree of haemorrhage/ Type of surgical procedure |
Factor VIII level required (%) |
Frequency of doses (hours)/Duration of therapy (days) |
|
Haemorrhage Early haemarthrosis, muscle bleeding or oral bleeding |
20 - 40 |
Repeat every 12 to 24 hours. At least 1 day, until the bleeding episode as indicated by pain is resolved or healing is achieved. |
|
More extensive haemarthrosis, muscle bleeding or haematoma |
30 - 60 |
Repeat every 12 to 24 hours for 3 - 4 days or more until pain and acute disability are resolved. |
|
Life threatening haemorrhages |
60 - 100 |
Repeat every 8 to 24 hours until threat is resolved. |
|
Surgery Minor surgery including tooth extraction |
30 - 60 |
Every 24 hours, at least 1 day, until healing is achieved. |
|
Major surgery |
80 - 100 (pre- and postoperative) |
Repeat every 8 to 24 hours until adequate wound healing, then therapy for at least another 7 days to maintain a factor VIII activity of 30 - 60%. |
Prophylaxis
For long term prophylaxis against bleeding in patients with severe haemophilia A, the usual doses are 20 to 40 IU of factor VIII per kg body weight at intervals of 2 to 3 days. In some cases, especially in younger patients, shorter dosage intervals or higher doses may be necessary.
Method of administration
Intravenous use. It is recommended not to administer more than 2-3 ml Haemoctin/min. For instructions on reconstitution of the medicinal product before administration, see section 6.6.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
4.4 Special warnings and precautions for use
Hypersensitivity
As with any intravenous protein product, allergic type hypersensitivity reactions are possible. The product contains traces of human proteins other than factor VIII. If symptoms of hypersensitivity occur, patients should be advised to discontinue use of the medicinal product immediately and contact their physician. Patients should be informed of the early signs of hypersensitivity reactions including hives, generalised urticaria, tightness of the chest, wheezing, hypotension, and anaphylaxis.
In case of shock, standard medical treatment for shock should be implemented.
Inhibitors
The formation of neutralising antibodies (inhibitors) to factor VIII is a known complication in the management of individuals with haemophilia A. These inhibitors are usually IgG immunoglobulins directed against the factor VIII procoagulant activity, which are quantified in Bethesda Units (BU) per ml of plasma using the modified assay. The risk of developing inhibitors is correlated to the exposure to factor VIII, this risk being highest within the first 20 exposure days. Rarely, inhibitors may develop after the first 100 exposure days.
Cases of recurrent inhibitor (low titre) have been observed after switching from one factor VIII product to another in previously treated patients with more than 100 exposure days who have a previous history of inhibitor development. Therefore, it is recommended to monitor all patients carefully for inhibitor occurrence following any product switch.
In general, all patients treated with human coagulation factor VIII products should be carefully monitored for the development of inhibitors by appropriate clinical observations and laboratory test. If the expected factor VIII activity plasma levels are not attained, or if bleeding is not controlled with an appropriate dose, testing for factor VIII inhibitor presence should be performed. In patients with high levels of inhibitor, factor VIII therapy may not be effective and other therapeutic options should be considered. Management of such patients should be directed by physicians with experience in the care of haemophilia and factor VIII inhibitors.
Cardiovascular events
In patients with existing cardiovascular risk factors, substitution therapy with factor VIII may increase the cardiovascular risk.
Catheter-related complications
If a central venous access device (CVAD) is required, risk of CVAD-related complications including local infections, bacteraemia and catheter site thrombosis should be considered.
Transmissible agents
Standard measures to prevent infections resulting from the use of medicinal products prepared from human blood or plasma include selection of donors, screening of individual donations and plasma pools for specific markers of infection and the inclusion of effective manufacturing steps for the inactivation/removal of viruses. Despite this, when medicinal products prepared from human blood or plasma are
administered, the possibility of transmitting infective agents cannot be totally excluded. This also applies to unknown or emerging viruses and other pathogens.
The measures taken are considered effective for enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus (HBV) and hepatitis C virus (HCV), and for the non-enveloped hepatitis A virus (HAV). The measures taken may be of limited value against non-enveloped viruses such as parvovirus B19.
Parvovirus B19 infection may be serious for pregnant women (foetal infection) and for individuals with immunodeficiency or increased erythropoiesis (e.g. haemolytic anaemia).
Appropriate vaccination (hepatitis A and B) should be considered for patients in regular/repeated receipt of human plasma-derived factor VIII products.
It is strongly recommended that every time Haemoctin is administered to a patient, the name and batch number of the product are recorded in order to maintain a link between the patient and the batch of the product.
Paediatric population
The special warnings and precautions for use mentioned for the adults should also be considered for the pediatric population.
Sodium content
One vial contains up to 32.2 mg sodium (1.4 mmol). To be taken into consideration by patients on a controlled sodium diet.
4.5 Interaction with other medicinal products and other forms of interaction
No interactions of human coagulation factor VIII products with other medicinal products have been reported.
4.6 Fertility, pregnancy and lactation
Animal reproduction studies have not been conducted with factor VIII. Based on the rare occurrence of haemophilia A in women, experience regarding the use of factor VIII during pregnancy and breast-feeding is not available. Therefore, factor VIII should be used during pregnancy and lactation only if clearly indicated.
4.7 Effects on ability to drive and use machines
Haemoctin has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
Hypersensitivity or allergic reactions (which may include angioedema, burning and stinging at the infusion site, chills, flushing, generalised urticaria, headache, hives, hypotension, lethargy, nausea, restlessness, tachycardia, tightness of the chest, tingling,
vomiting, wheezing) have been observed rarely and may in some cases progress to severe anaphylaxis (including shock).
Patients with haemophilia A may develop neutralising antibodies (inhibitors) to factor VIII. If such inhibitors occur, the condition will manifest itself as an insufficient clinical response. In such cases, it is recommended that a specialised haemophilia centre be contacted.
For safety information with respect to transmissible agents, see section 4.4.
Tabulated list of adverse reactions
The table presented below is according to the MedDRA system organ classification (SOC and Preferred Term Level).
Frequencies have been evaluated according to the following convention: very common (>1/10); common (>1/100 to <1/10); uncommon (>1/1,000 to <1/100); rare (>1/10,000 to <1/1,000); very rare (<1/10,000), not known (cannot be estimated from the available data).
From clinical trials, non interventional studies, spontaneous reporting and regular literature screening the following adverse reactions were reported on Haemoctin 250, 500 and 1000:
|
MedDRA Standard System Organ Class |
Adverse reactions |
Frequency |
|
Skin and subcutaneous tissue disorder |
Exanthema, urticaria, erythema |
very rare |
|
Investigations |
Anti factor VIII antibody positive |
very rare |
Paediatric population
Frequency, type and severity of adverse reactions in the paediatric population are expected to be the same as in adults.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system: Yellow Card Scheme, Website: www.mhra.gov.uk/yellowcard.
4.9 Overdose
No case of overdose has been reported.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic Group: antihaemorrhagics: blood coagulation factor VIII. ATC code: B02BD02.
The factor Vlll/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions. When infused into a haemophiliac patient, factor VIII binds to von Willebrand factor in patients circulation.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X to activated factor X (factor Xa). Activated factor X converts prothrombin into
thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed. Haemophilia A is a sex-linked hereditary disorder of blood coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints, muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By replacement therapy the plasma levels of factor VIII are increased, thereby enabling a temporary correction of the factor deficiency and correction of the bleeding tendencies.
In addition to its role as a factor VIII protecting protein, von Willebrand factor mediates platelet adhesion to sites of vascular injury and plays a role in platelet aggregation.
Data on successfully performed Immune Tolerance Induction (ITI) have been collected in patients with haemophilia A who have developed inhibitors to factor VIII.
5.2 Pharmacokinetic properties
Plasma factor VIII activity decreases by a two-phase exponential decay after intravenous use. In the initial phase, distribution between intravascular and other compartments (body fluids) occurs with a half-life of elimination from the plasma of 1 to 8 hours. In the subsequent phase the half-life varies between 5 - 18 hours, with an average of about 12 hours. This appears to correspond to the true biological half-life.
The incremental recovery of Haemoctin is approximately 0.020 ± 0.003 IU/ml/IU/kg b.w. The level of factor VIII activity after intravenous use of 1 IU factor VIII per kg b.w. is about 2 %.
Other pharmacokinetic parameters of Haemoctin are:
• Area under the curve (AUC): about 17 IU x h / ml
• Mean residence time (MRT): about 15 h
• Clearance: about 155 ml/h.
5.3 Preclinical safety data
Human plasma coagulation factor VIII (from the concentrate) is a normal constituent of the human plasma and acts like the endogenous factor VIII. Single dose toxicity testing is of no relevance since higher doses result in overloading. Repeated dose toxicity testing in animals is impracticable due to the interference with developing antibodies to heterologous protein.
Even doses of several times the recommended human dose per kilogram body weight show no toxic effects on laboratory animals.
Since clinical experience provides no hint for tumorigenic and mutagenic effects of human plasma coagulation factor VIII, experimental studies, particularly in heterologous species, are not considered imperative.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Powder: glycine, sodium chloride, sodium citrate, calcium chloride Solvent: water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
Only the provided infusion sets should be used because treatment failure can occur as a consequence of human coagulation factor VIII adsorption to the internal surfaces of some infusion equipment.
6.3 Shelf life
2 years
After first opening, the product should be used immediately.
6.4 Special precautions for storage
Do not store above 25 C. Do not freeze.
Keep the vials in the outer carton in order to protect from light.
6.5 Nature and contents of container
1 package Haemoctin contains:
1 vial with powder (20 ml) out of glass type I acc. to Ph. Eur. Freeze-drying stoppers out of halobutyl-caoutchouc, type I acc. to Ph. Eur.
1 vial with solvent (5 ml, 10 ml), glass type I acc. to Ph. Eur. Injection stoppers out of halobutyl-caoutchouc, type I acc. to Ph. Eur.
The pack also contains:
1 disposable syringe (5 ml, 10 ml), 1 transfer system with integral filter, 1 butterfly cannula
6.6 Special precautions for disposal
Reconstituted medicinal product should be inspected visually for particulate matter and discoloration prior to administration. The solution should be clear or slightly opalescent. Do not use solutions that are cloudy or have deposits.
Any unused product or waste material should be disposed of in accordance with local requirements.
Instructions for use and handling:
Absolute sterility is to be ensured in all steps of the procedure!
Fig. 1
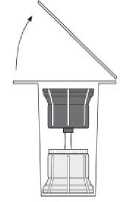
Fig. 2

Fig. 5
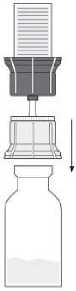
Fig. 4
Fig. 6
Dissolution of the concentrate:
• Bring the unopened vials of the solvent (water for injections) and product to room temperature. If a water bath is used for warming, it must be scrupulously ensured that the water does not come into contact with the caps or stoppers of the vials. Otherwise contamination of the medicine may occur.
• Remove the caps from both vials in order to expose the central portions of the rubber stoppers (1). Ensure that the rubber stoppers of the product and solvent vials are treated with a disinfectant.
• Remove the top of the transfer system packaging (2) Place the blue part of the transfer system onto the upright standing vial containing the solvent (3).
• Remove the remaining part of the packaging of the transfer system. Now the transparent part of the transfer system is visible.
• Place the product vial on an even surface.
• Turn the combination of transfer system and solvent vial upside down. Push the spike of the transparent part of the adapter straight down through the product vial stopper (4). The vacuum present in the product vial causes the solvent to flow into the product vial. (5) Immediately unscrew the blue part of the transfer system together with the solvent vial. Discard the solvent vial with the blue part of the transfer system attached (6). Gently swirling the product vial helps in dissolving the powder. Do not shake vigorously, all foaming is to be avoided! The solution is clear or slightly opalescent.
• The solution ready for use should be used immediately after dissolving. Do not use solutions that are cloudy or contain visible particles.

Fig. 7
Injection:
• Once you have dissolved the powder as described above, screw the enclosed syringe with its Luer-Lock connector onto the product vial with the transparent part of the transfer system. (7) This allows you to easily draw the dissolved drug into the syringe. A separate filter is not necessary because the transfer system has its own integral filter.
• Carefully disconnect the vial with the transparent part of the transfer system from the syringe. Use the enclosed butterfly needle and administer immediately by slow intravenous injection. The injection rate must not exceed 2-3 ml/minute.
• After the butterfly needle has been used, it can be made safe with the protective cap.
7 MARKETING AUTHORISATION HOLDER
Biotest Pharma GmbH, Landsteinerstrasse 5, 63303 Dreieich, Germany tel: (49) 6103 801-0 fax: (49) 6103 801-150
8 MARKETING AUTHORISATION NUMBER(S)
PL 04500/0009 PL 04500/0010
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
06/08/2008 / 04/09/2008
10 DATE OF REVISION OF THE TEXT
16/09/2016