Lutrate 1 Month Depot 3.75 Mg Powder And Solvent For Prolonged-Release Suspension For Injection
1.
NAME OF THE MEDICINAL PRODUCT
Lutrate 1 month Depot 3.75 mg powder and solvent for prolonged-release suspension for injection.
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each vial contains 3.75 mg of leuprorelin acetate (equivalent to 3.57 mg leuprorelin free base).
1 ml of reconstituted suspension contains 1.875 mg of leuprorelin acetate.
Excipients with known effect:
Each vial contains from 1.3 to 2.2 mg (<1 mmol) of sodium (as carmellose sodium).
For the full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Powder and solvent for prolonged-release suspension for injection.
Powder: white to off-white powder.
Solvent: clear transparent solution (pH 5.0 - 7.0).
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
Lutrate 1 month Depot is indicated for palliative treatment of locally advanced or metastatic prostate
cancer.
4.2 Posology and method of administration
Posology
The usual recommended dose of Lutrate 1 month Depot is 3.75 mg presented as a one month depot injection and administered as a single intramuscular injection every month.
Lutrate 1 month Depot must be administered under the supervision of a physician or a qualified health practitioner.
The dose of Lutrate 1 month Depot allowing the continuous release of leuprorelin acetate during one month is incorporated in a depot formulation. The lyophilized powder should be reconstituted and administered as a single intramuscular injection at monthly intervals. Intra-arterial or intravenous administration must be avoided. The vial of Lutrate 1 month Depot microsphere powder should be reconstituted immediately prior to administration by intramuscular injection. As with other drugs administered regularly by injection, the injection site should be varied periodically.
Lutrate 1 month Depot therapy should not be discontinued when remission or improvement occurs.
Response to Lutrate 1 month Depot therapy should be monitored measuring serum levels of testosterone as well as prostate-specific antigen (PSA) periodically. Clinical studies have shown that testosterone levels increased during the first 4 days of treatment in the majority of non-orchiectomised patients. They then decreased and reached castrate levels by 3-4 weeks. Once attained, castrate levels (defined as testosterone less than 0.5 ng/mL) were maintained as long as drug therapy continued.
If a patient's response appears to be sub-optimal, then it would be advisable to confirm that serum testosterone levels have reached or are remaining at castrate levels. Transient increases in acid phosphatase levels sometimes occur early in the treatment period but usually return to normal or near normal values by the 4th week of treatment.
Duration of treatment
Lutrate 1 month Depot has been administered as monthly intramuscular injections.
Special populations
Paediatric population
The safety and efficacy of Lutrate 1 month Depot in the paediatric patients has not been established. Therefore, Lutrate 1 month Depot is not recommended in children or adolescents until safety and efficacy data become available.
Patients with renal/hepatic impairment
The pharmacokinetics of Lutrate 1 month Depot in hepatically and renally impaired patients has not been determined.
Older people
In the clinical trial for Lutrate 1 month Depot, the mean age of the subjects studied was 71.6±9.2 years. Therefore, the labelling reflects the pharmacokinetics, efficacy and safety of Lutrate 1 month Depot in this population.
Method of Administration
Lutrate 1 month Depot must be administered via the intramuscular route only. Do not administer by any other route. If it is administered subcutaneously by mistake, the patient should be closely monitored since no data about other administration routes a part from intramuscular is available for Lutrate 1 month Depot For instructions on reconstitution of the medicinal product before administration, see section 6.6.
4.3 Contraindications
Hypersensitivity to the active substance, luteinising hormone releasing hormone (LHRH) analogs or any of the excipients listed in section 6.1. Reports of anaphylactic reactions to synthetic LHRH or LHRH agonist analogs have been reported in the medical literature.
Previous orchiectomy.
Lutrate 1 month Depot must not be used as the only treatment in patients with prostate cancer and with evidence of spinal cord compression or spinal metastases.
Lutrate 1 month Depot is not indicated for use in women.
Lutrate 1 month Depot is not indicated for use in paediatric patients.
4.4 Special warnings and precautions for use
In the initial stages of Lutrate 1 month Depot treatment, as occurs during treatment with other LHRH agonists, a transient rise in levels of testosterone may occur. In some cases, this may be associated with a "flare" or exacerbation of the tumour growth resulting in temporary worsening of prostate cancer symptoms. These symptoms usually subside on continuation of therapy (see section 4.8).. "Flare" may manifest itself as systemic or neurological symptoms in some cases (i.e. bone pain...). Also, cases of orchiatrophy and gynecomastia have been described with other LHRH agonists.
Treatment should be discontinued immediately if the patient develops any signs or symptoms suggestive of anaphylaxis/anaphylactic reaction (dyspnoea, asthma, rhinitis, angioneurotic oedema or glottis, hypotension, urticaria, rash, pruritus or interstitial pneumonitis). Patients should be informed before starting treatment, warning them to discontinue it and consult their doctor if any of the above mentioned symptoms occur. Patients who have experienced a hypersensitivity reaction to leuprorelin should be closely monitored and should not be rechallenged with Lutrate 1 month Depot.
In patients treated with leuprorelin acetate, isolated cases of urethral obstruction (with or without haematuria) and spinal cord compression or metastatic vertebral lesions have been observed, which may contribute to paralysis with or without fatal complications. Patients at risk of urethral obstruction, spinal cord compression or metastatic vertebral lesions should be considered carefully and closely supervised in the first few weeks of treatment. These patients should be considered for prophylactic treatment with anti-androgens.
Should urological/neurological complications occur, these should be treated by appropriate specific measures.
There is an increased risk of incident depression (which may be severe) in patients undergoing treatment with GnRH agonists, such as leuprorelin acetate. Patients should be informed accordingly and treated as appropriate if symptoms occur.
Decreased bone density has been reported in the medical literature in men who have had orchiectomy or who have been treated with an LHRH agonist. Adding antiandrogenic therapy to the treatment regimen reduces bone loss, but increases the risk of other adverse effects such as clotting problems and oedema. If an antiandrogen is used over a prolonged period, due attention should be paid to the contraindications and precautions associated with its extended use. Patients at risk or with a medical history of osteoporosis should be considered carefully and closely supervised during treatment with leuprorelin acetate (see section 4.8).
Hepatic dysfunction and jaundice with elevated liver enzyme levels have been reported with the use of leuprorelin acetate. Therefore, close observation should be made and appropriate measures taken if necessary.
Response to Lutrate 1 month Depot therapy should be monitored by clinical parameters and by measuring testosterone and PSA serum levels periodically.
Patients may experience metabolic changes (e.g. glucose intolerance or worsening of existing diabetes), hypertension, weight changes, and cardiovascular disorders. As would be expected with this class of drug, development or aggravation of diabetes may occur, therefore diabetic patients may require more frequent monitoring of blood glucose during treatment with Lutrate 1 month Depot. Patients at high risk for metabolic or cardiovascular diseases should be carefully assessed before commencing treatment and adequately monitored during androgen deprivation therapy. Therapy with leuprorelin acetate results in suppression of the pituitary-gonadal system. Results of diagnostic tests of pituitary gonadotropic and gonadal functions conducted during and after leuprorelin acetate therapy may be affected.
Increased prothrombin time has been reported in patients under treatment with leuprorelin acetate.
Seizures have been reported with the administration of leuprorelin acetate. These cases were observed in patients with a history of seizures, epilepsy, cerebrovascular disorders, anomalies or central nervous system tumours and in patients with concomitant medications that have been associated with seizures for example bupropion and selective inhibitors of serotonin reuptake (SSRIs). Seizures in patients in the absence of the any medical conditions mentioned above have also been reported.
Leuprorelin acetate should be used with precautions in the presence of, cardiovascular disease (including congestive heart failure condition), thromboembolism, oedema, depression, and pituitary apoplexy.
Leuprorelin acetate should be used with caution in patients with known bleeding disorders, thrombocytopenia or on treatment with anticoagulants. Sportsmen should take precaution as Lutrate 1 month Depot contains an ingredient which may give a positive test result in doping controls.
This medicinal product contains less than 1 mmol sodium (23 mg) per vial. It is essentially ‘sodium-free’.
Androgen deprivation therapy may prolong the QT interval.
In patients with a history of or risk factors for QT prolongation and in patients receiving concomitant medicinal products that might prolong the QT interval (see section 4.5) physicians should assess the benefit risk ratio including the potential for Torsade de pointes prior to initiating Lutrate 1 month Depot.
4.5 Interaction with other medicinal products and other forms of interaction
No pharmacokinetic-based drug-drug interaction studies have been conducted with leuprorelin acetate. However, because leuprorelin acetate is a peptide that is primarily degraded by peptidase and not by Cytochrome P-450 enzymes as noted in specific studies, and the drug is only about 46% bound to plasma proteins, pharmacokinetic drug interactions would not be expected to occur.
Since androgen deprivation treatment may prolong the QT interval, the concomitant use of Lutrate 1 month Depot with medicinal products known to prolong the QT interval or medicinal products able to induce Torsade de pointes such as class IA (e.g. quinidine, disopyramide) or class III (e.g. amiodarone, sotalol, dofetilide, ibutilide) antiarrhythmic medicinal products, methadone, moxifloxacin, antipsychotics, etc. should be carefully evaluated (see section 4.4).
4.6 Fertility, pregnancy and lactation
Pregnancy:
Lutrate 1 month Depot is not indicated for use in pregnant women.
Leuprorelin acetate injection may cause foetal harm when administered to a pregnant woman.
Therefore, the possibility exists that spontaneous abortion may occur if the drug is administered during pregnancy.
Breastfeeding:
Lutrate 1 month Depot should not be used in women who are breastfeeding.
Fertility:
Studies in animals have shown reproductive toxicity (see section 5.3).
4.7 Effects on ability to drive and use machines
No specific studies on the effects of Lutrate 1 month Depot on the ability to drive and use machines have been performed. However, the ability to drive and use machines may be impaired due to visual disturbances and dizziness.
4.8 Undesirable effects
Unless otherwise specified, the following safety profile of Lutrate 1 month Depot is based on the results of a phase III clinical trial in which prostate cancer patients were treated with six intramuscular monthly doses of Lutrate 1 month Depot and followed up for total a period of 26 weeks. Most of the treatment-related AEs reported were the usual ones associated with testosterone suppressing therapy.
The most commonly reported adverse reactions with Lutrate 1 month Depot are hot flushes, injection site pain, injection site irritation, night sweats and headache.
The following adverse reactions from clinical investigations were listed below by system organ class and in order of decreasing incidence (very common: >1/10; common: >1/100 to <1/10; uncommon: > 1/1,000 to < 1/100; rare: >1/10,000 to <1/1,000; very rare: < 1/10,000).
Category
SOC
Frequency: PT
Metabolism and nutrition disorders
Common: Increased appetite
Uncommon: Anorexia, hypercholesterolaemia, hyperlipidaemia
Psychiatric disorders
Uncommon: Sleep disorders, insomnia, libido decreased, mood changes and depression*
Nervous system disorders
Common: Headache Uncommon: Somnolence
Ear and labyrinth disorders
Uncommon: Vertigo
Vascular disorder
Very common: Hot flush
Gastrointestinal disorders
Uncommon: Abdominal pain lower, diarrhoea, nausea, vomiting
Hepatobiliary disorders
Uncommon: Hyperbilirubinaemia
Skin and subcutaneous tissue disorders
Common: Hyperhidrosis, night sweats, cold sweats
_Uncommon: Periorbital edema, urticaria, pruritus,_
Musculoskeletal and connective tissue disorders
Common: Back pain
_Uncommon: Arthralgia, muscle spasms, pain in extremity_
Renal and urinary disorders
Uncommon: Urinary retention, urinary incontinence, pollakiuria
Reproductive system and breast disorders
Common: Erectile dysfunction
_Uncommon: Breast swelling, breast tenderness, ejaculation failure_
General disorders and administration site conditions
Common: Fatigue, asthenia, pyrexia, local adverse reactions (see table 2)
_Uncommon: Weakness, feeling hot and cold, feeling jittery_
Investigations
Uncommon: Increased AST, increased ALT, bilirubin increased, gamma
_glutamyltransferase increased, electrocardiogram QT prolonged (see sections 4.4 and 4.5).
* In a post-marketing study the frequency of mood changes and depression in long term users was common.
In terms of severity, 98% of all treatment-related AEs were mild or moderate. Eighty-nine percentage (89%) of the hot flushes were reported as mild and nine percentage (9%) as moderate. Two cases of hot flushes (0.2%) were reported as severe.
A total of 35 local adverse reactions (LAR) at the injection site were reported by 29 patients (18.1%) during the study.
Local adverse reactions after Lutrate 1 month Depot 3.75mg are those typically reported with other similar products administered via intramuscular injection. Injection site pain, injection site irritation, injection site discomfort, injection site bruising and erythema were the most commonly reported. Uncommonly reported reactions were injection site reaction, swelling, injury and haemorrhage (Table 2)
Table 2. Frequency of patients with local adverse reactions during Lutrate 1 month Depot therapy.
Primary SOC* Patients with related LAR
PT: General disorders and administration site %
conditions
|
Common | |
|
Injection site pain |
8.1 |
|
Injection site irritation |
4.4 |
|
Injection site discomfort |
1.9 |
|
Injection site erythema |
1.3 |
|
Injection site bruising |
1.3 |
Uncommon Injection site reaction Injection site swelling Injection site injury 0.6
0.6
0.6
Injection site haemorrhage 0.6
*Subjects may fall into more than one category; LAR: local adverse reaction; SOC: System Organ Class.
In the presence of repeated administrations of Lutrate 1 month Depot, swelling (0.6%) , pain (0.6%), bruising (0.6%) and irritation (0.6%) were reported as recurrent local adverse reactions. These events were all reported as not serious and mild. No patient discontinued therapy due to local adverse events.
In a phase I clinical trial (CRO-02-43) carried out in healthy subjects with Leuprolide Depot GP-Pharm 7.5 mg administered at single dose, one case of injection site induration was reported.
Other adverse events which have been reported to occur with leuprorelin acetate treatment include impotence, decrease in libido (both pharmacological consequences of testosterone deprivation), peripheral oedema, pulmonary embolism, palpitations, myalgia, muscle weakness, chills, dyspnoea, peripheral vertigo, rash, amnesia, visual disturbances and skin sensation. Infarction of pre-existing pituitary adenomas has been reported rarely after administration of both short and long acting LHRH agonists. There have been rare reports of thrombocytopenia and leucopoenia. Changes in glucose tolerance have been reported.
4.9 Overdose
There is no clinical experience with the effects of an acute overdose of Lutrate 1 month Depot or leuprorelin acetate. In clinical trials using daily subcutaneous leuprorelin acetate in patients with prostate cancer, doses as high as 20 mg/day for up to two years caused no AEs differing from those observed with the 1 mg/day dose.
In animal studies, doses of up to 500 times the recommended human dose resulted in dyspnoea, decreased activity and local irritation at the injection site. In cases of overdosage, the patient should be monitored closely and management should be symptomatic and supportive.
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Endocrine therapy. Hormones and related agents. Gonadotropin-releasing hormones analogues; ATC code: L02AE02.
The chemical name of Leuprorelin acetate is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-L-prolyl-ethylamide.
Leuprorelin acetate is inactive when given orally due to poor membrane permeability and an almost complete inactivation by intestinal proteolytic enzymes.
Leuprorelin acetate has potent LHRH agonist properties when given during short-term and intermittent therapy, however, when administered in a continuous, nonpulsatile manner, LHRH analogs induce inhibition of gonadotropin secretion and suppression of testicular steroidogenesis.
Upon binding to pituitary LHRH receptors, leuprorelin acetate produces an initial increase in circulating levels of luteinizing hormone (LH) and follicle stimulating hormone (FSH), leading to an acute rise in levels of testosterone and dihydrotestosterone. However, within five to eight days after drug administration, LHRH analogs produce desensitization of the LHRH receptor complex and/or downregulation of the anterior pituitary gland. Due to the fact that there are fewer receptors on the cell surface, cellular stimulation is decreased, and less gonadotropin is synthesized and secreted. Eventually, after several weeks of LHRH agonist therapy, LH and FSH secretion is suppressed. As a result, Leydig cells in the testes cease to produce testosterone, and the serum testosterone concentration declines to a castration level (less than 0.5 ng/mL) in about two to four weeks after initiation of treatment.
In an open-label, multicenter, multiple dose clinical study of Lutrate 1 month Depot, 160 patients with prostate cancer and no previous systemic cancer therapy, hormonal therapy for the treatment of prostate cancer, previous prostatic surgery neither previous orchiectomy, were enrolled. The objectives were to determine the efficacy and safety of Lutrate 1 month Depot when given to prostate cancer patients who could benefit from androgen deprivation therapy. Lutrate 1 month Depot was administered intramuscularly in 6 monthly doses.
Testosterone levels were monitored at different days during 168 days. As expected, after the first injection the mean testosterone levels rapidly increased from baseline levels (4.119± 1.341 ng/mL), reaching peak levels (Cmax) of 6.598±2.249 ng/mL at the third day. After peaking, testosterone levels fell, and by day 21, 78.7% of the evaluable patients had achieved medical castration (defined as testosterone less than 0.5 ng/mL). By Day 28, 96.8% of the patients had achieved castrate levels, and 73.1% had reached levels of <0.2 ng/mL (Figure 1).
Figure 1. Mean (±SD) testosterone plasma levels during treatment with six monthly IM injections of Lutrate 1 month Depot 3.75 mg
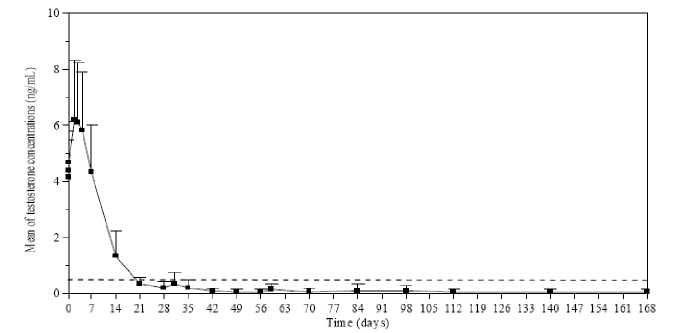
Secondary efficacy endpoints included determination of serum LH, FSH and PSA concentrations. By day 14 and day 4 after the first Lutrate 1 month Depot injection, mean LH and FSH serum levels had decreased below the baseline concentrations. Concentrations remained well below baseline values from day 28 until the end of the study. During the treatment, mean PSA serum levels gradually decreased (first month) and then remained constantly below baseline level until the end of the study. However, a wide inter-individual variation in PSA concentrations was observed throughout the study.
The frequency of the acute on chronic response was 10.5 % and the frequency of testosterone breakthrough response was 11.8 %. No drug-related adverse events suggestive of a clinical testosterone flare (urinary retention, spinal cord compression, or exacerbation of bone pain) were reported in any of the patients showing a testosterone breakthrough effect.
5.2 Pharmacokinetic properties
Absorption
Following three once-monthly injections of Lutrate 1 month Depot in a sample of prostate cancer patients (N=12), maximal leuprorelin acetate plasma concentration was similar among the three cycles. After first administration (Days 0-28), Cmax was 13,145.6±3,070.6 pg/ml. Median time to achieve Cmax (Tmax) was 0.04 days, corresponding to 0.96 h (range 0.96 - 4.08 h).
Distribution
No drug distribution study was conducted with Lutrate 1 month Depot. However, in healthy male volunteers, the mean steady-state volume of distribution of leuprorelin acetate following bolus intravenous (IV) 1.0 mg dose was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Elimination
No drug metabolism or excretion study was conducted with Lutrate 1 month Depot.
Leuprorelin is expected to be metabolised to smaller inactive peptides that may be excreted or further catabolised.
In healthy male volunteers, a 1.0 mg bolus of leuprorelin acetate administered IV revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
Following administration of leuprorelin acetate to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
Special Populations
Patients with renal/hepatic impairment
The pharmacokinetics of the drug in hepatically and renally impaired patients has not been determined.
5.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity and genotoxicity conducted with leuprorelin acetate.
As expected from its known pharmacological properties, non-clinical studies showed effects on the reproductive systems, which were reversible. In the reproductive toxicity studies, leuprorelin acetate did not show teratogenicity. However, embryotoxicity/lethality was observed in rabbits.
Carcinogenicity studies performed in rats with leuprorelin acetate administered subcutaneously (0.6 to 4 mg/kg/day), showed a dose-related increase in pituitary adenomas Furthermore a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males was observed the highest incidence was in the low dose group. Administration of leuprorelin acetate resulted in inhibition of the growth of certain hormone dependent tumours (prostatic tumours in Noble and Dunning male rats and DMBA-induced mammary tumours in female rats). No such effects were observed in carcinogenicity studies performed in mice. No carcinogenicity studies have been conducted with Lutrate 1 month Depot.
Studies with leuprorelin acetate showed that the product was not mutagenic in a set of in vitro and in vivo assays. No mutagenicity studies have been conducted with Lutrate 1 month Depot.
6 PHARMACEUTICAL PARTICULARS 6.1 List of excipients
Excipients of the lyophilizate (vial):
Polysorbate 80 Mannitol (E-421)
Carmellose sodium (E-466)
Triethyl citrate
Poly(DL-lactide-co-glycolide) (PLGA)
Excipients of the solvent (prefilled syringe):
Mannitol (E-421)
Sodium hydroxide (for pH adjustment)
Hydrochloric acid (for pH adjustment)
Water for injection
6.1 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
No other solvent other than the sterile solvent provided for Lutrate 1 month Depot can be used for the reconstitution of Lutrate 1 month Depot powder.
6.3 Shelflife
3 years unopened.
Once reconstituted with the solvent the suspension should be administered immediately.
6.4 Special precautions for storage
Do not store above 25o C. Do not freeze.
Store in the original package in order to protect from light.
For storage conditions after reconstitution of the medicinal product, see section 6.3.
6.5 Nature and contents of container
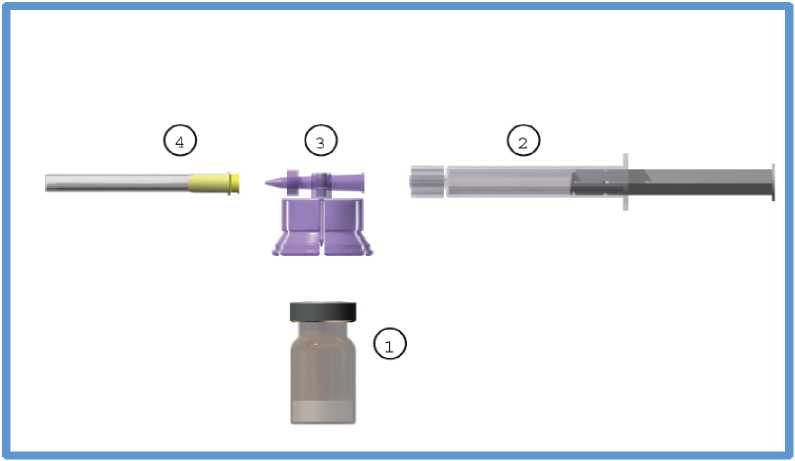
The commercial kit includes:
1. One (1) type I glass vial containing 3.75 mg of leuprorelin acetate as a freeze-dried powder, sealed with a bromobutyl stopper and an aluminium flip-off cap.
2. One (1) type I glass prefilled syringe containing 2 ml of solvent sealed with an elastomer cap.
3. One (1) polycarbonate / HDPE adaptor system including one (1) sterile 20 gauge needle.
6.2 Special precautions for disposal and other handling
Method of administration
The vial of Lutrate 1 month Depot microsphere powder should be reconstituted immediately prior to administration by intramuscular injection. Make sure an aseptic technique is followed.
The reconstituted product is a suspension of milky, white colour appearance.
No other solvent can be used for reconstitution Lutrate 1 month Depot.
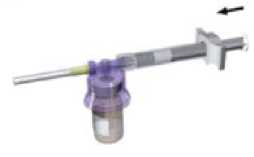
Reconstitute Lutrate 1 month Depot according to the following instructions:

syringe and attach it to the adaptor system
4
While keeping the syringe and vial securely coupled in an upright position, slowly push the plunger in order to transfer all the diluents into the vial
7
5
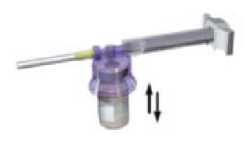
With the syringe still coupled to the vial, shake the vial gently for approximately one minute until a uniform milky-white suspension is obtained

Detach the syringe and needle from the adaptor system by twisting the upper piece of the adaptor counterclockwise. The drug is ready to be used.
6

Turn the system upside down, and carefully pull out the plunger to draw up the resuspended drug from the vial into the syringe
8

Clean the injection area with an alcohol swab and let the skin dry. Inject the suspension intramuscularly into the upper outer quadrant of the gluteus
Some product may cake or clump at the vial wall. This is considered normal. During product manufacture the vial is filled with excess product in order to make sure that a final dose of 3.75 mg of leuprorelin acetate is administered.
The product is meant for a single injection. Any remaining suspension must be discarded.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
Mercury Pharmaceuticals Limited Capital House, 1st Floor,
85 King William Street,
London EC4N 7BL,
United Kingdom
8 MARKETING AUTHORISATION NUMBER(S)
PL 12762/0509
9. DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
Date of first authorization: 16 June 2015
10 DATE OF REVISION OF THE TEXT
06/10/2015