Numeta G16%E Emulsion For Infusion
SUMMARY OF PRODUCT CHARACTERISTICS
This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions.
1 NAME OF THE MEDICINAL PRODUCT
Numeta G16%E emulsion for infusion
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
This medicinal product is presented in the form of a three chamber bag. Each bag contains a sterile non-pyrogenic combination of a glucose solution, a paediatric amino acids solution, with electrolytes, and a lipid emulsion, as described below.
|
Container size |
50% glucose solution |
5.9% amino acids solution with electrolytes |
12.5% lipid emulsion |
|
500 mL |
155 mL |
221 mL |
124 mL |
If lipid administration is undesirable, the design of the bag allows the possibility to activate only the peel seal between the amino acids/electrolytes and glucose chambers, leaving the peel seal between the amino acids and lipid chambers intact. The content of the bag can subsequently be infused with or without lipids. The composition of the drug product after mixing of the two (amino acids and glucose, 2 chamber bag, 376 mL solution) or three (amino acids, glucose and lipid, 3 chamber bag, 500 mL emulsion) chambers are provided in the following table.
|
Composition | ||
|
Active Substance |
Activated 2CB |
Activated 3CB |
|
(376 mL) |
(500 mL) | |
|
Amino Acid Chamber | ||
|
Alanine |
1.03 g |
1.03 g |
|
Arginine |
1.08 g |
1.08 g |
|
Aspartic acid |
0.77 g |
0.77 g |
|
Cysteine |
0.24 g |
0.24 g |
|
Glutamic acid |
1.29 g |
1.29 g |
|
Glycine |
0.51 g |
0.51 g |
|
Histidine |
0.49 g |
0.49 g |
|
Isoleucine |
0.86 g |
0.86 g |
|
Leucine |
1.29 g |
1.29 g |
|
Lysine monohydrate |
1.59 g |
1.59 g |
|
(equivalent to Lysine) |
(1.42 g) |
(1.42 g) |
|
Methionine |
0.31 g |
0.31 g |
|
Ornithine hydrochloride |
0.41 g |
0.41 g |
|
(equivalent to Ornithine) |
(0.32 g) |
(0.32 g) |
|
Phenylalanine |
0.54 g |
0.54 g |
|
Proline |
0.39 g |
0.39 g |
|
Serine |
0.51 g |
0.51 g |
|
Taurine |
0.08 g |
0.08 g |
|
Threonine |
0.48 g |
0.48 g |
|
Tryptophan |
0.26 g |
0.26 g |
|
Tyrosine |
0.10 g |
0.10 g |
|
Valine |
0.98 g |
0.98 g |
|
Sodium chloride |
0.30 g |
0.30 g |
|
Potassium acetate |
1.12 g |
1.12 g |
|
Calcium chloride dihydrate |
0.46 g |
0.46 g |
|
Magnesium acetate tetrahydrate |
0.33 g |
0.33 g |
|
Sodium glycerophosphate hydrated |
0.98 g |
0.98 g |
|
Glucose Chamber | ||
|
Glucose monohydrate |
85.25 g |
85.25 g |
|
(equivalent to glucose anhydrous) |
(77.50 g) |
(77.50 g) |
|
Lipid Chamber | ||
|
Refined olive oil (approximately 80%) + Refined soya bean oil (approximately 20%) |
- |
15.5 g |
2CB=two chamber bag, 3CB=three chamber bag
For the full list of excipients, see section 6.1.
The reconstituted solution/emulsion provides the following:
|
Composition | ||||
|
Activated 2CB |
Activated 3CB | |||
|
Per volume unit (mL) |
376 |
100 |
500 |
100 |
|
Nitrogen (g) |
2.0 |
0.52 |
2.0 |
0.39 |
|
Amino acids (g) |
13.0 |
3.5 |
13.0 |
2.6 |
|
Glucose (g) |
77.5 |
20.6 |
77.5 |
15.5 |
|
Lipids (g) Energy |
0 |
0 |
15.5 |
3.1 |
|
Total calories (kcal) |
362 |
96 |
517 |
103 |
|
Non-protein calories (kcal) |
310 |
82 |
465 |
93 |
|
Glucose calories (kcal) |
310 |
82 |
310 |
62 |
|
Lipid calories (kcal)a |
0 |
0 |
155 |
31 |
|
Non-prot calories / nitrogen (kcal/g N) |
158 |
158 |
237 |
237 |
|
Lipid calories (%non-protein calories) |
NA |
N/A |
33 |
33 |
|
Lipid calories (% total calories) Electrolytes |
NA |
N/A |
30 |
30 |
|
Sodium (mmol) |
11.6 |
3.1 |
12.0 |
2.4 |
|
Potassium (mmol) |
11.4 |
3.0 |
11.4 |
2.3 |
|
Magnesium (mmol) |
1.6 |
0.41 |
1.6 |
0.31 |
|
Calcium (mmol) |
3.1 |
0.82 |
3.1 |
0.62 |
|
Phosphateb (mmol) |
3.2 |
0.85 |
4.4 |
0.87 |
|
Acetate (mmol) |
14.5 |
3.9 |
14.5 |
2.9 |
|
Malate (mmol) |
4.3 |
1.1 |
4.3 |
0.86 |
|
Chloride (mmol) |
13.8 |
3.7 |
13.8 |
2.8 |
|
pH (approx.) |
5.5 |
5.5 |
5.5 |
5.5 |
|
Osmolarity approx. (mOsm/L) |
1585 |
1585 |
1230 |
1230 |
a Includes calories from egg phosphatide,
b Includes phosphate from egg phosphatide component of the lipid emulsion
3 PHARMACEUTICAL FORM
Emulsion for infusion.
Appearance before reconstitution:
• The solutions in the amino acids and glucose chambers are clear, colourless or slightly yellow
• The lipid emulsion is homogeneous and milky-white
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Numeta G16%E is indicated for parenteral nutrition in term newborn infants and children up to 2 years when oral or enteral nutrition is not possible, insufficient or contraindicated.
4.2 Posology and method of administration
Posology
The dosage depends on energy expenditure, the patient’s weight, age, clinical status, and on the ability to metabolize the constituents of Numeta, as well as on additional energy or proteins given orally/enterally. Total electrolyte and macronutrient composition is dependent on the number of activated chambers (See section 2).
The maximum daily dose should not be exceeded. Due to the static composition of the multi-chamber bag, the ability to simultaneously meet all nutrient needs of the patient may not be possible. Clinical situations may exist where patients require amounts of nutrients varying from the composition of the static bag.
The maximal recommended hourly rate of infusion and volume per day depend on the constituent. The first of these limits to be reached sets the maximum daily dose. The guidelines for maximal recommended hourly rate of infusion and volume per day are:
|
Activated 2CB (376 mL) |
Activated 3CB (500 mL) | |
|
Maximal rate of infusion in mL/kg/h |
5.8 |
5.5 |
|
Corresponding to: | ||
|
Amino acid in g/kg/h |
0.20a |
0.14 |
|
Glucose in g/kg/h |
1.2 |
0.85 |
|
Lipids in g/kg/h |
0 |
0.17a |
|
Maximal amount in mL/kg/day |
72.3 |
96.2 |
|
Corresponding to: | ||
|
Amino acid in g/kg/d |
2.5a |
2.5a |
|
Glucose in g/kg/d |
14.9 |
14.9 |
|
Lipids in g/kg/d |
0 |
3.0 |
Limiting parameter according to ESPEN-ESPGHAN guidelines
Method of administration
For instructions for preparation, and handling of the solution/emulsion for infusion, see section 6.6.
Due to its high osmolarity, undiluted Numeta can only be administered through a central vein. However, sufficient dilution of Numeta with water for injection lowers the osmolarity and allows peripheral infusion. The table below indicates how the dilution impacts osmolarity of the bags.
Examples of osmolarity for activated 2CB and activated 3CB admixtures after addition of trace elements, vitamins, and water for injection:
|
Amino Acids and Glucose (Activated 2CB) |
Amino Acids, Glucose, and Lipids (Activated 3CB) | |
|
Initial volume in the bag (mL) |
376 |
500 |
|
Initial osmolarity (mOsm/L approximately) |
1585 |
1230 |
|
Volume of water added (mL) |
450 |
350 |
|
Trace elements added a |
5 ml TE2 |
5 ml TE2 |
|
Vitamins added a |
vial V1 |
vial V1 + vial V2 |
|
Final volume after addition (mL) |
836 |
860 |
|
Osmolarity after addition (mOsm/L approximately) |
715 |
715 |
a Composition of vitamins and trace elements preparations are described below.
Composition of the commercial trace elements preparation used
|
Composition per vial |
TE2 (10 mL) |
|
Iron |
8.9pmol or 0.5mg |
|
Zinc |
15.3pmol or 1mg |
|
Selenium |
0.6pmol or 0.05mg |
|
Copper |
4.7pmol or 0.3mg |
|
Iodine |
0.4pmol or 0.05mg |
|
Fluorine |
26.3pmol or 0.5mg |
|
Molybdenum |
0.5pmol or 0.05mg |
|
Manganese |
1.8pmol or 0.1mg |
|
Cobalt |
2.5pmol or 0.15mg |
|
Chromium |
0.4pmol or 0.02mg |
Composition of the commercial vitamin preparations used
|
Composition per vial |
V1 |
V2 |
|
Vitamin B1 |
25mg |
- |
|
Vitamin B2 |
3.6mg |
- |
|
Nicotinamide |
40mg |
- |
|
Vitamin B6 |
4.0mg |
- |
|
Pantothenic acid |
15.0mg |
- |
|
Biotin |
60pg |
- |
|
Folic acid |
400pg |
- |
|
Vitamin B12 |
50hg |
- |
|
Vitamin C |
100mg |
- |
|
Vitamin A |
- |
2300IU |
|
Vitamin D |
- |
400IU |
|
Vitamin E |
- |
7IU |
|
Vitamin K |
- |
200pg |
The flow rate should be increased gradually during the first hour. Upon discontinuation of Numeta, the flow rate should be decreased gradually during the last hour. The administration flow rate must be adjusted taking into account the dose being administered, the daily volume intake, and the duration of the infusion, see section 4.9.
The same bag should not be activated, hung, and infused longer than 24 hours. Cyclic infusions should be managed according to the patient’s metabolic tolerance.
Treatment with parenteral nutrition may be continued for as long as is required by the patient’s clinical conditions.
This product contains electrolytes and may be further supplemented using commercial electrolyte preparations according to the physician’s judgment and the clinical needs of the patient, see section 6.6.
Vitamins and trace elements can be added according to the physician’s judgment and the clinical needs of the patient, see section 6.6.
4.3 Contraindications
The general contraindications for administering Numeta as an activated 2 chamber bag for intravenous infusion are as follows:
• Hypersensitivity to egg, soy or peanut proteins, or to any of the active substances, excipients listed in section 6.1, or components of the container
• Congenital abnormality of the amino acid metabolism
• Pathologically elevated plasma concentrations of sodium, potassium, magnesium, calcium and/or phosphorus
• As for other calcium-containing infusion solutions, concomitant treatment with ceftriaxone in newborn infants (<28 days of age), even if separate infusion lines are used (risk of fatal ceftriaxone calcium salt precipitation in the neonate's bloodstream). See sections 4.4, 4.5 and 6.2.
• Severe hyperglycaemia
The addition of lipids (administering Numeta as an activated 3 chamber bag for intravenous emulsion) is contraindicated in the following additional clinical situations:
• Severe hyperlipidaemia, or severe disorders of lipid metabolism characterized
by hypertriglyceridemia
4.4 Special warnings and precautions for use
The infusion must be stopped immediately if any signs or symptoms of an allergic reaction (such as fever, sweating, shivering, headache, skin rashes, or dyspnea) develop.
Cases of fatal reactions with calcium-ceftriaxone precipitates in lungs and kidneys in full-term newborns aged less than 1 month have been described.
In patients of any age ceftriaxone must not be mixed or administered simultaneously with intravenous calcium-containing solutions, including Numeta, .even via different infusion lines or at different infusion sites because of the risk of precipitation of ceftriaxone-calcium salt.
However, in patients older than 28 days of age ceftriaxone and calcium-containing solutions may be administered sequentially one after another if infusion lines at different sites are used or if the infusion lines are replaced or thoroughly flushed between infusions with physiological salt-solution to avoid precipitation.
Pulmonary vascular precipitates causing pulmonary vascular embolism and respiratory distress have been reported in patients receiving parenteral nutrition. In some cases, fatal outcomes have occurred. Excessive addition of calcium and phosphate increases the risk of the formation of calcium phosphate precipitates (see section 6.2). Suspected precipitate formation in the blood stream have also been reported.
In addition to inspection of the solution, the infusion set and catheter should also periodically be checked for precipitates.
If signs of respiratory distress occur, the infusion should be stopped and medical evaluation initiated.
No additions to the bag should be made without first checking the compatibility, as formation of precipitates or destabilization of the lipid emulsion could result in vascular occlusion, see sections 6.2 and 6.6.
Infection and sepsis may occur as a result of the use of intravenous catheters to administer parenteral formulations, or poor maintenance of catheters. Immunosuppressive effects of illness, or drugs, may promote infection and sepsis. Careful symptomatic and laboratory monitoring for fever/chills, leukocytosis, technical complications with the access device, and hyperglycaemia can help recognize early infections. Patients who require parenteral nutrition are often predisposed to infectious complications due to malnutrition and/or their underlying disease state. The occurrence of septic complications can be decreased with
heightened emphasis on aseptic technique in catheter placement, maintenance, as well as aseptic technique in nutritional formula preparation.
Fat overload syndrome has been reported with other parenteral nutrition products. The reduced or limited ability to metabolize the lipids contained in Numeta may result in a “fat overload syndrome”.
Refeeding severely undernourished patients may result in the refeeding syndrome that is characterized by the shift of potassium, phosphorus, and magnesium intracellularly as the patient becomes anabolic. Thiamine deficiency and fluid retention may also develop. Careful and slow initiation of parenteral nutrition is recommended, with close monitoring of fluids, electrolytes, trace elements and vitamins.
Numeta must only be administered through a central vein, except if appropriate dilution is performed (see section 4.2). When making additions to the formulation, the final osmolarity of the mixture must be calculated before administration via peripheral vein to avoid vein irritation.
Do not connect bags in series in order to avoid air embolism due to possible residual gas contained in the primary bag.
Lipids, vitamins, additional electrolytes and trace elements should be administered as required.
PRECAUTIONS
Do not add other medicinal products or substances to one of the three chambers of the bag or to the reconstituted solution/emulsion without first confirming their compatibility and the stability of the resulting preparation (in particular, stability of the lipid emulsion) (see sections 6.2 and 6.6).
Routinely monitor water and electrolyte balance, serum osmolarity, serum triglycerides, acid/base balance, blood glucose, liver and kidney function, blood count including platelets, and coagulation parameters throughout treatment.
In case of unstable conditions (for example, following severe post-traumatic conditions, uncompensated diabetes mellitus, acute phase of circulatory shock, acute myocardial infarction, severe metabolic acidosis, severe sepsis and hyperosmolar coma) delivery of Numeta should be monitored and adjusted to meet the clinical needs of the patient.
Cardiovascular
Use with caution in patients with pulmonary edema or heart failure. Fluid status should be closely monitored.
Renal
Use with caution in patients with renal insufficiency. Fluid and electrolyte status including magnesium (see Hypermagnesaemia) should be closely monitored in these patients.
Severe water and electrolyte equilibration disorders, severe fluid overload states, and severe metabolic disorders should be corrected before starting the infusion (see section 4.3).
Hepatic/Gastrointestinal
Use with caution in patients with severe liver insufficiency, including cholestasis, or elevated liver enzymes. Liver function parameters should be closely monitored.
Endocrine and Metabolism
Metabolic complications may occur if the nutrient intake is not adapted to the patient's requirements, or the metabolic capacity of any given dietary component is not accurately assessed. Adverse metabolic effects may arise from administration of inadequate or excessive nutrients or from inappropriate composition of an admixture for a particular patient's needs.
Serum triglyceride concentrations and the ability of the body to metabolize lipids must be checked regularly. If a lipid metabolism abnormality is suspected, monitoring of serum triglycerides is recommended as clinically necessary.
In the event of hyperglycemia, the infusion rate of Numeta must be adjusted and/or insulin administered, see section 4.9.
Hematologic
Use with caution in patients with severe blood coagulation disorders. Blood count and coagulation parameters should be closely monitored.
Hypermagnesaemia
Numeta G16%E provides 0.3 mmol/kg/d of magnesium when administered at maximum dose (see section 4.2). There is a possibility that this may lead to hypermagnesaemia. The signs of hypermagnesaemia include generalised weakness, hypo-reflexia, nausea, vomiting, hypocalcaemia, respiratory failure, hypotension and arrhythmias. As signs of hypermagnesaemia may not be detected, monitoring of magnesium levels is advised at baseline and at appropriate intervals thereafter, in accordance with routine clinical practice and the needs of the individual patient. This is especially important in those patients at increased risk of developing hypermagnesaemia including patients with impaired renal function, patients receiving other medicinal products which place them at risk of developing hypermagnesaemia or patients receiving magnesium from other sources, including neonates whose mother’s recently received magnesium in the antepartum period.
If serum magnesium levels are elevated (above reference range normal values) the infusion of Numeta should be stopped or infusion rate reduced as deemed clinically appropriate and safe.
4.5 Interaction with other medicinal products and other forms of interaction
No pharmacodynamic interaction studies have been performed with Numeta.
Numeta must not be administered simultaneously with blood through the same infusion tubing because of the risk of pseudoagglutination.
As for other calcium-containing infusion solutions concomitant treatment with ceftriaxone and Numeta G16%E is contraindicated in term newborn infants (<28 days of age), even if separate infusion lines are used (risk of fatal ceftriaxone-calcium salt precipitation in the neonate’s bloodstream).
In patients of any age (including adults), ceftriaxone must not be mixed or administered simultaneously with any intravenous calcium-containing solutions, including Numeta, even via different infusion lines or at different infusion sites because of the risk of precipitation of ceftriaxone-calcium salt (see section 4.4).
However, in patients older than 28 days of age ceftriaxone and calcium-containing solutions may be administered sequentially one after another if infusion lines at different sites are used or if the infusion lines are replaced or thoroughly flushed between infusions with physiological salt-solution to avoid precipitation.
Olive and soybean oil have a natural content of vitamin K1 that may counteract the anticoagulant activity of coumarin (or coumarin derivatives including warfarin).
Due to the potassium content of Numeta special care should be taken in patients simultaneously treated with potassium sparing diuretics (amiloride, spironolactone, triamterene) or with ACE inhibitors, angiotensin II receptor antagonists, or the immunosuppressants tacrolimus and cyclosporine in view of the risk of hyperkalemia.
The lipids contained in this emulsion may interfere with the results of certain laboratory tests (for example, bilirubin, lactate dehydrogenase, oxygen saturation, blood hemoglobin) if the blood sample is taken before the lipids are eliminated.
Lipids are generally eliminated after a period of 5 to 6 hours when no additional lipids are administered.
Please also refer to section 6.2 “Incompatibilities”.
4.6 Fertility, pregnancy and lactation
Not relevant
4.7 Effects on ability to drive and use machines
Not relevant
4.8 Undesirable effects
The safety and administration of Numeta was assessed in a single phase III study. One hundred and fifty nine (159) paediatric patients were included in the study and received Numeta.
The following table summarizes the Adverse Reactions seen in this study
|
Clinical Trial Adverse Reactions | ||
|
System Organ Class (SOC) |
Preferred MedDRA Term |
Frequencyb |
|
METABOLISM AND NUTRITION DISORDERS |
Hypophosphataemia3 |
Common |
|
Hyperglycaemia3 |
Common | |
|
Hypercalcaemiaa |
Common | |
|
Hypertriglyceridaemiaa |
Common | |
|
Hyperlipidaemia |
Uncommon | |
|
Hyponatraemiaa |
Common | |
|
HEPATOBILIARY DISORDERS |
Cholestasis |
Uncommon |
a Blood samples drawn during the infusion (without fasting conditions). b Frequency is based upon the following categories: Very Common (>1/10); Common (>1/100 -<1/10), Uncommon (>1/1,000 - <1/100), Rare (>1/10,000 - <1/1,000), Very Rare (<1/10,000).
The following adverse reactions have been reported with other parenteral nutrition admixtures:
Fat overload syndrome: may be caused by inappropriate administration (e.g. overdose and/or infusion rate higher than recommended, see section 4.9); however the signs and symptoms of this syndrome may also occur when the product is administered according to instructions. The reduced or limited ability to metabolize the lipids contained in Numeta accompanied by prolonged plasma clearance may result in a “fat overload syndrome”. This syndrome is associated with a sudden deterioration in the patient’s clinical condition and is characterized by findings such as hyperlipidemia, fever, liver fatty infiltration (hepatomegaly), deteriorating liver function, anemia, leukopenia, thrombocytopenia, coagulation disorders and central nervous system manifestations (e.g. coma). The syndrome is usually reversible when the infusion of the lipid emulsion is stopped.
Pulmonary vascular precipitates (pulmonary vascular embolism and respiratory distress) (see section 4.4).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme.
Website: www.mhra.gov.uk/yellowcard
4.9 Overdose
In the event of inappropriate administration (overdose, and/or infusion rate higher than recommended), nausea, vomiting, shivering, electrolyte disturbances and signs of hypervolaemia or acidosis may occur and result in fatal consequences. In such
situations, the infusion must be stopped immediately. If medically appropriate, further intervention may be indicated.
Hyperglycaemia, glucosuria, and hyperosmolar syndrome may develop if the glucose infusion rate exceeds clearance.
The reduced or limited ability to metabolize lipids may result in fat overload syndrome, the results of which are usually reversible after infusion of the lipid emulsion is stopped, see section 4.8.
There is no specific antidote for overdose. Emergency procedures should be general supportive measures, with particular attention to respiratory and cardiovascular systems. In some serious cases, hemodialysis, hemofiltration, or hemodiafiltration may be necessary.
Close biochemical monitoring is essential and specific abnormalities should be treated appropriately.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Solutions for parenteral nutrition/combination
ATC Code: B05 BA10
The content of nitrogen (20 L-series amino acids, including 8 essential amino acids) in NUMETA and energy (glucose and triglycerides) enables maintenance of an adequate nitrogen/energy balance. Nitrogen and energy are required for normal functioning of all cells in the body, and are important for protein synthesis, growth, wound healing, immune function, muscle function, and many other cellular activities.
This formulation also contains electrolytes.
The amino acids profile is as follows:
• Essential amino acids/total amino acids: 47.5%
• Branched-chain amino acids/total amino acids: 24.0%
The lipid emulsion included in Numeta is a mixture of refined olive oil and refined soybean oil (ratio 80/20 approximately), with the following relative distribution of fatty acids:
• 15% saturated fatty acids (SFA)
• 65% monounsaturated fatty acids (MUFA)
• 20% polyunsaturated fatty acids (PUFA)
The phospholipid/triglyceride ratio is 0.06. The moderate essential fatty acid (EFA) content improves the status of their upper derivatives while correcting EFA deficiency.
Olive oil contains significant amounts of alpha-tocopherol which, when combined with a moderate PUFA intake, contributes to vitamin E status and is important for limiting lipid peroxidation.
The carbohydrate source is glucose. Glucose is a primary source of energy in the body.
5.2 Pharmacokinetic properties
The ingredients of the emulsion for infusion (amino acids, electrolytes, glucose, lipids) are distributed, metabolized and eliminated in the same way as if they had been administered individually. The product is given intravenously and is thus 100% bioavailable and the constituents are distributed to and metabolized by all cells in the body.
5.3 Preclinical safety data
Preclinical studies performed on the components of the triple chamber bag have revealed no additional risks to those already mentioned in other sections of the SmPC.
Animal studies with Numeta (double or triple chamber combinations) have not been conducted.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
|
Excipients: |
Amino acid Chamber |
Glucose Chamber |
Lipid Chamber |
|
L-Malic acid a |
X |
- |
- |
|
Hydrochloric acid a |
- |
X |
- |
|
Purified egg phosphatide |
- |
- |
X |
|
Glycerol |
- |
- |
X |
|
Sodium oleate |
- |
- |
X |
|
Sodium hydroxide a |
- |
- |
X |
|
Water for injections |
X |
X |
X |
|
a for pH adjustment |
6.2 Incompatibilities
Do not add other medicinal products or substances to one of the three components of the bag or to the reconstituted solution/emulsion without first confirming their compatibility and the stability of the resulting preparation (in particular, stability of the lipid emulsion or formation of precipitates), see section 6.6.
As with any parenteral nutrition admixture, calcium and phosphate ratios must be considered. Excess addition of calcium and phosphate, especially in the form of mineral salts may result in the formation of calcium phosphate precipitates.
In patients of any age ceftriaxone must not be mixed or administered simultaneously with intravenous calcium-containing solutions, including Numeta,_even via different
infusion lines or at different infusion sites because of the risk of precipitation of ceftriaxone-calcium salt.
Numeta must not be administered simultaneously with blood through the same infusion tubing, see section 4.5.
Numeta contains calcium ions which pose additional risk of coagulation precipitated in citrate-anticoagulated/preserved blood or components.
6.3 Shelf life
18 months
Shelf life after reconstitution
It is recommended that the product be used immediately after the non-permanent seals between the two or three chambers have been opened. However stability data of the reconstituted mixtures supports 7 days between 2°C and 8°C followed by 48 hours at 30°C.
Shelf life after supplementation (electrolytes, trace elements, vitamins, water):
For specific admixtures in-use stability of the Numeta formulation has been demonstrated for 7 days between 2°C and 8°C followed by 48 hours at 30°C.
From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2 to 8°C, unless reconstitution /dilution /supplementation has taken place in controlled and validated aseptic conditions.
Please also refer to section 4.2 “Posology and method of administration” and section 6.6 “Special precautions for disposal and other handling”.
6.4 Special precautions for storage
Do not freeze.
Store in overpouch.
6.5 Nature and contents of container
The three-chamber full non-PVC bag consists of the following components:
• A multi-layer plastic sheeting
• A port tube on the compartment containing the lipid emulsion. It is sealed off after filling to prevent additions to this chamber.
• Two port tubes on the amino acid solution and glucose solution chambers.
• An injection site that closes the port tube of the glucose compartment.
• An administration site that closes the port tube of the amino acid compartment.
All components are free of natural latex rubber.
To prevent air contact, the bag is packaged in an oxygen barrier overpouch that contains an oxygen absorber sachet and may also contain an oxygen indicator.
Available pack sizes:
500 mL bags: 6 units per cardboard box
1 bag of 500 mL
Not all pack sizes may be marketed
6.6 Special precautions for disposal
For single use only.
It is recommended that after the non-permanent seals between the chambers have been opened, the contents should be used immediately, and should not be stored for subsequent infusion.
Do not connect bags in series in order to avoid air embolism due to possible residual gas contained in the primary bag.
Confirm the integrity of the bag and of the non-permanent seals. Use only if the bag is not damaged, if the non-permanent seals are intact (i.e., no content mixture of any of the three chambers), if the solution in the amino acids chamber and the solution in the glucose chamber are clear, colorless, or slightly yellow, practically free of visible particles, and if the lipid emulsion is a homogeneous liquid with a milky appearance.
Before opening the overpouch, check the color of the oxygen indicator. Compare it to the reference color printed next to the OK symbol and depicted in the printed area of the indicator label. Do not use the product if the color of the oxygen indicator does not correspond to the reference color printed next to the OK symbol.
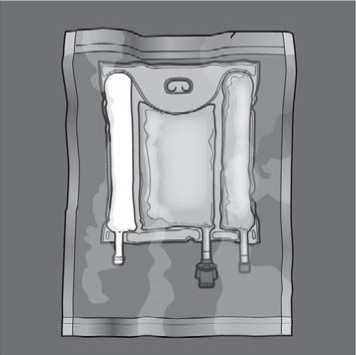
To open: Remove the protective overpouch. Discard the overpouch and oxygen absorber/indicator sachet.
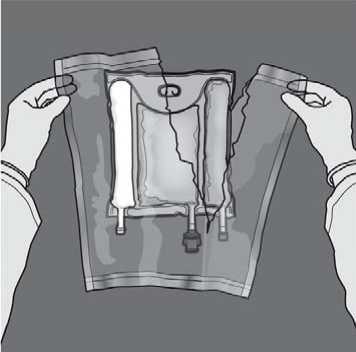
Mixing: Ensure that the product is at room temperature when breaking the nonpermanent seals. Place the bag onto a flat clean surface. Do not use the bag if the contents are mixed due to accidental rupture of compartment seals during transport.
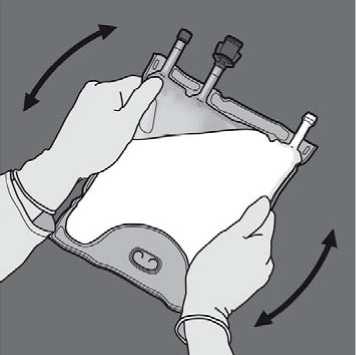
Activation of the 3CB (breaking two non-permanent seals)
Start rolling the bag from the D-hanger side.
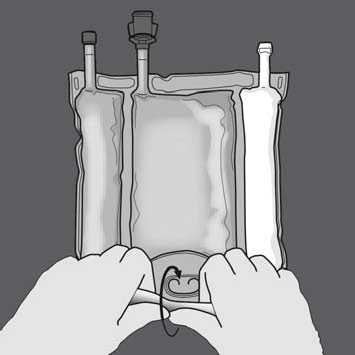
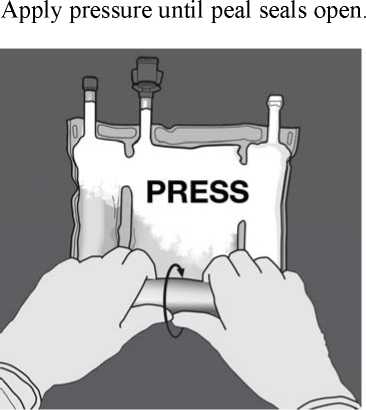
Then change direction by rolling the bag towards the D-hanger, continue until the seal is completely opened
Proceed the same way to complete the opening of the second peel seal.
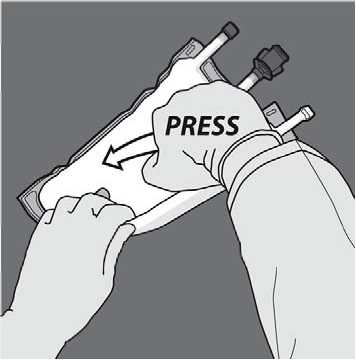
Turn the bag over at least three times to mix the contents thoroughly. The appearance of the mixed solution should be a milky-white emulsion.
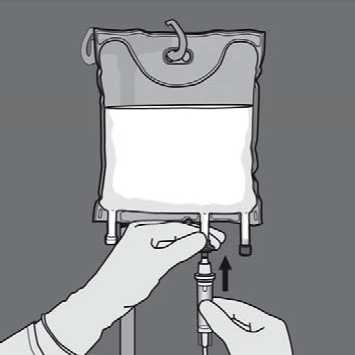
Remove the protective cap from the administration site and insert the IV administration set.
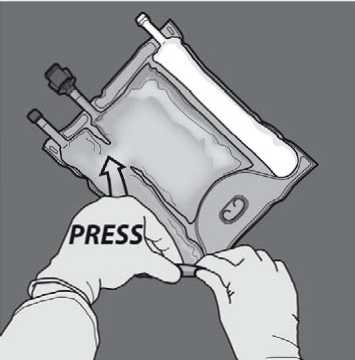
Activation of the 2CB (breaking the non-permanent seal between the Amino Acids and Glucose chambers)
To break only the amino acids/glucose peel seal, start rolling the bag from the D-hanger corner of the seal separating the amino acids and glucose chambers and apply pressure to open the seal separating the glucose and amino acids compartments.
Orient the bag such that the lipid emulsion compartment is nearest to the operator and roll the bag while protecting the lipid emulsion compartment in the palms of the hands.
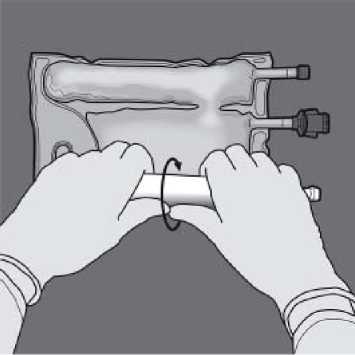
With one hand, apply pressure by rolling the bag towards the tubes.
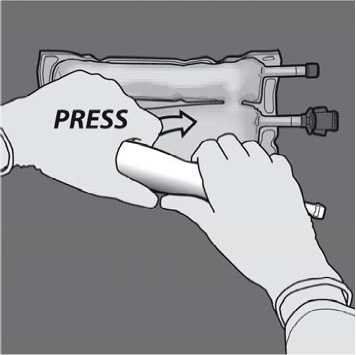
Then change direction by rolling the bag towards the D-hanger, pressing with the other hand, continuing until the seal separating the amino acids and glucose solutions is completely opened.
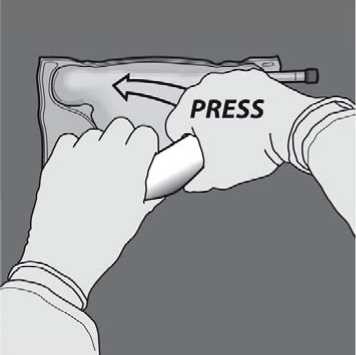
Turn the bag over at least three times to mix the content thoroughly. The appearance of the mixed solution should be clear, colorless or slightly yellow.
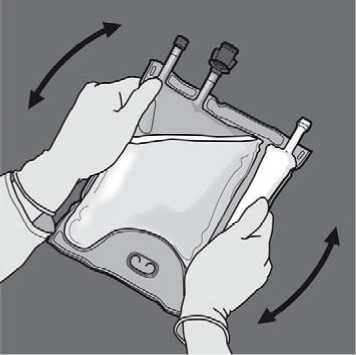
Remove the protective cap from the administration site and insert the IV administration set.
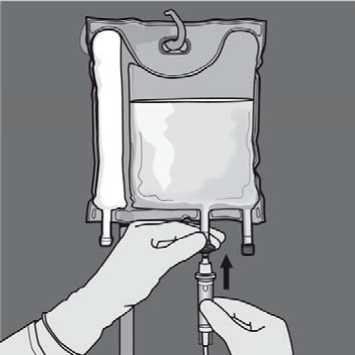
Additions: The capacity of the bag is sufficient to enable additions such as, electrolytes, trace elements, vitamins and water for injection. Any addition (including vitamins) may be made into the reconstituted mixture (after the non-permanent seals have been opened and after the contents of the two or three chambers have been mixed). Vitamins may also be added into the glucose chamber before the mixture is reconstituted (before opening the non-permanent seals and before mixing the solutions and the emulsion).
When making additions, the amount of electrolytes already present in the bag should be taken into account to meet the clinical needs of the patient. Maximum levels of electrolytes, and commercially available vitamins and trace elements formulations that can be added to Numeta activated 2CB and activated 3CB admixtures are described below:
|
Additive |
Maximal Further Addition Per Bag | |
|
Amino Acids and Glucose |
Amino Acids, Glucose, and Lipids | |
|
(Activated 2 CB) |
(Activated 3 CB) | |
|
Sodium |
26.0 mmol |
4.0 mmol |
|
Potassium |
26.2 mmol |
6.2 mmol |
|
Magnesium |
3.6 mmol |
0.0 mmol |
|
Calcium |
8.2 mmol |
2.1mmol |
|
Phosphatea |
8.1 mmol |
2.0 mmol |
|
Trace elements b |
5 mL TE1 |
5 mL TE1 |
|
Or 5 mL TE2 |
Or 5 mL TE2 | |
|
Vitamins b |
A vial V1 |
A vial V1 + A vial V2 |
a: Organic phosphate not including phosphate from egg phosphatide.
b: Composition of trace elements (i.e. TE2) and vitamins (i.e. V1 & V2) preparations are illustrated in Section 4.2. Composition of trace elements (i.e. TE1) is described below.
Composition of the commercial trace elements preparation used
|
Composition per vial |
TE1 (10 mL) |
|
Zinc |
38.2 pmol or 2.5 mg |
|
Selenium |
0.253 pmol or 0.02 mg |
|
Copper |
3.15 pmol or 0.2 mg |
|
Iodine |
0.0788 pmol or 0.01 mg |
|
Fluorine |
30 pmol or 0.57 mg |
|
Manganese |
0.182 pmol or 0.01 mg |
To perform an addition:
• Aseptic conditions must be observed
• Prepare the injection site of the bag
• Puncture the injection site and inject the additives using an injection needle or a reconstitution device
• Mix content of the bag and the additives Preparation of the infusion:
• Aseptic conditions must be observed
• Suspend the bag
• Remove the plastic protector from the administration outlet
• Firmly insert the infusion set spike into the administration outlet Administration of the infusion:
• For single use only
• Only administer the product after the non-permanent seals between the two or three chambers have been opened and the contents of the two or three chambers have been mixed
• Ensure that the final activated 3CB emulsion for infusion does not show any evidence of phase separation or the final 2CB solution for infusion does not show any evidence of particles
• After opening the bag, the content must be used immediately, and never stored for a subsequent infusion
• Do not connect any partially used bag
• Do not connect in series in order to avoid the possibility of air embolism due to possible residual gas contained in the primary bag
Any unused product or waste material and all necessary disposable devices must be properly discarded and not used again.
MARKETING AUTHORISATION HOLDER
Baxter Healthcare Limited Caxton Way,
Thetford,
Norfolk,
IP24 3SE,
United Kingdom
8
9
MARKETING AUTHORISATION NUMBER(S)
PL 00116/0648
DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
08/04/2016
DATE OF REVISION OF THE TEXT
08/04/2016