Plenaxis 100 Mg Powder And Solvent For Suspension For Injection
Out of date information, search anotherSUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Plenaxis 100 mg powder and solvent for suspension for injection.
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each individual dose contains 100 mg of abarelix.
After reconstitution with 2.2 ml of sodium chloride solution, the suspension for injection contains 50 mg/ml of abarelix.
This medicinal product contains sodium and is administered in sodium chloride 9 mg/mL (0.9%) solution for injection (see section 6.6). After reconstitution, the solution contains less than 1 mmol of sodium (23 mg). To be taken into consideration by patients on a controlled sodium diet.
For a full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Powder and solvent for suspension for injection. A white powder and a clear, colourless solution. Intramuscular use.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Plenaxis is indicated in adult male patients for the initiation (up to 85 days) of hormonal castration in advanced or metastasising hormone-dependent prostate carcinoma when androgen suppression is necessary.
4.2 Posology and method of administration
Posology
Plenaxis 100 mg should be administered by intramuscular injection (i.m.) on Day 1, Day 15, Day 29 (Week 4) and subsequently every 4 weeks. The
success of Plenaxis therapy can be monitored by means of clinical parameters and the periodic measurement of serum testosterone and PSA levels. An increased PSA should be confirmed with a check of serum testosterone before diagnosing a castrate refractory prostate cancer.
Particular attention should be paid to the possible decrease in efficacy in the case of patients with a body weight > 100 kg.
Caution is advised in mild to moderate hepatic or renal impairment since data are insufficient to provide specific dosing recommendations. Plenaxis should not be used in severe hepatic or renal impairment.
Plenaxis may be substituted with a GnRH agonist following initiation of treatment. Data from 176 patients indicate that substitution of Plenaxis with a GnRH agonist was undertaken after 3 months without a clinically significant increase in testosterone levels (testosterone surge or flare).
Paediatric population
Plenaxis should not be used in children.
Method of administration
Plenaxis must be reconstituted prior to administration.
For instructions on reconstitution of the medicinal product before administration, see section 6.6.
Plenaxis should be administered by intramuscular injection.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients.
Plenaxis should not be administered to patients with severe hepatic or renal impairment.
4.4 Special warnings and precautions for use
Plenaxis must not be used in children until data in this population become available.
Plenaxis must not be used in women.
Patients should be kept under observation for at least 30 minutes after every Plenaxis injection and, in the event of an allergic immediate-type reaction, adequate measures must be instituted (e.g. epinephrine, corticosteroids, antihistamines, oxygen, administration of intravenous fluids and/or raising the legs, either as individual measures or in combination) (see also Section 4.8).
Since Plenaxis may prolong the QT interval in ECGs, the doctor should weigh carefully the risks involved in using Plenaxis against the benefits of treatment in patients whose basic QTc values are >450 milliseconds (e.g. in congenital QT prolongation), or who are receiving Class Ia anti-arrhythmics (e.g. chinidine, procainamide) or Class III anti-arrhythmics (e.g. amiodarone, sotalol) and/or other drugs known to prolong QT, which therefore have an additive effect, such as haloperidol, methadone, paliperdone, pantamidine, cisaprid, venlafaxine or vorinostat (See Section 4.8).
During clinical studies with Plenaxis, a small group of patients had to be excluded from the study, because they showed a permanent rise in transaminase values. Serum transaminase values should therefore be ascertained both at the beginning of treatment with Plenaxis and periodically during treatment. If either the AST value or the ALT value (SGOT, SGPT), or both values, are more than 3 times as high as the upper normal limit value, the values should be ascertained for a second time before administration of the next dose of Plenaxis. If the second value is still more than twice as high as the upper normal limit value, the patient should be excluded from further treatment with Plenaxis. If the second measured value is less than twice as high as the upper normal limit value, the patient may continue to be treated, but the ascertainment of transaminase values should be planned for the following month and then regularly every month after that. In clinical trials, most cases of raised levels of transaminase were transient and became normal again during the course of treatment.
Plenaxis should not be administered to patients who show restricted hepatic and/or renal function, until new data in this population become available.
Particular attention should be paid to the possible decrease in efficacy described in Section 4.2 in the case of patients with a body weight > 100 kg.
Decreased bone density has been reported in the medical literature in men who have had orchiectomy or who have been treated with a GnRH agonist. It can be anticipated that long periods of testosterone suppression in men will have effects on bone density. Bone density has not been measured during treatment with abarelix.
A reduction in glucose tolerance has been observed in men who have had orchiectomy or who have been treated with a GnRH agonist. Development or aggravation of diabetes may occur; therefore diabetic patients may require more frequent monitoring of blood glucose when receiving androgen deprivation therapy. The effect of abarelix on insulin and glucose levels has not been studied.
Plenaxis contains sodium, but less than 1 mmol (23 mg) of sodium per dosage unit. This should be taken into consideration by patients on a controlled sodium diet.
4.5 Interaction with other medicinal products and other forms of interaction
No interaction studies have been performed. No interaction can be anticipated with medicinal products metabolised by cytochrome P-450 enzymes, because these enzymes do not have any effect on Plenaxis metabolism. Plenaxis shows a very high percentage of plasma protein binding (96-99%). Patients treated with other medicinal products also showing a high percentage of plasma protein binding could have raised serum concentrations of these medicinal products, which might result in the occurrence of side-effects.
No changes in coagulation parameters (PT and PTT) have been observed in patients taking warfarin during treatment with Plenaxis.
Since Plenaxis may prolong the QT interval in ECGs, the doctor should weigh carefully the risks involved in using Plenaxis against the benefits of treatment in patients whose basic QTc values are >450 milliseconds (e.g. in congenital QT prolongation), or who are receiving Class Ia anti-arrhythmics (e.g. chinidine, procainamide) or Class III anti-arrhythmics (e.g. amiodarone, sotalol) and/or other drugs known to prolong QT, which therefore have an additive effect, such as haloperidol, methadone, paliperdone, pantamidine, cisaprid, venlafaxine or vorinostat (See Section 4.8).
4.6 Pregnancy and lactation
Plenaxis is not indicated for use in women (see Section 4.1).
Non-clinical data reveal no special hazard for humans based on conventional toxicity to reproduction studies. Abarelix is embryolethal in rats and rabbits. It cannot be excluded that abarelix is also teratogenic. Most of the malformations occurred in the abarelix-treated groups but data were inconclusive as there were too few litters and foetuses available for adequate evaluation.
Plenaxis reduces circulating levels of FSH, LH and testosterone. In male patients Plenaxis is likely to result in reversible decreases in libido, reversible erectile dysfunction and reduction in fertility. Plenaxis is not indicated in women but would be expected to reversibly reduce with fertility and pregnancy. Plenaxis should not be administered to children or adolescents and is likely to inhibit sexual maturation.
4.7 Effects on ability to drive and use machines
Plenaxis has no or negligible influence on the ability to drive and use machines. Fatigue and dizziness immediately following administration of Plenaxis are common adverse reactions thought to be related to the actual injection or the underlying disease. However, patients will be observed in the clinic for 30 minutes following administration so Plenaxis should have no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Immediate-type systemic allergic reactions (e.g. urticaria, pruritus, hypotension and/or syncopes) occurred in the case of 1.1% of patients (15 out of 1,357) treated with Plenaxis. The symptoms appeared within 30 minutes of administration of the medicinal product in all 15 patients. Of these 15 patients, 7 suffered hypotension and/or fainting as part of their immediate-type systemic allergic reactions.
The cumulative risk of an immediate-type systemic allergic reaction increased with the duration of treatment with Plenaxis.
The cumulative rates for all immediate-type systemic allergic reactions on Days 56, 141, 365 and 675 after the initial dose were 0.51%, 0.80%, 1.24% and 2.91% respectively. For the sub-group of patients who suffered hypotension and/or fainting as part of their immediate-type systemic allergic reactions, the cumulative rates on Days 56, 141, 365 and 675 after the initial dose were 0.22%, 0.32%, 0.61% and 1.67% respectively.
The undesirable effects corresponded to those anticipated in medically castrated prostatic cancer patients with additional accompanying diseases. Pain, respiratory tract infections, frequent gastrointestinal disturbances, urinary flow disturbances because of the underlying tumour, as well as the known symptoms of medical castration were among the most frequently reported undesirable effects.
Frequency definitions: very common (> 1/10), common (> 1/100 to < 1/10), uncommon (> 1/1000 to < 1/100), rare (> 1/10000 to < 1/1000) and very rare (> 1/10000).
Adverse effects are categorised by system organ class and frequency.
The most frequent adverse reactions are:
|
System Organ Class |
Very Common (>1/10) |
Common (>1/100 to <1/10) |
Uncommon (>1/1,000 to <1/100) |
|
Blood and lymphatic system disorders |
Coagulopathy (coagulation abnormal), anaemia. | ||
|
Cardiac disorders |
Heart enlarged, arrhythmia, ventricular arrhythmia, bradycardia, tachycardia, palpitations, coronary artery disease. | ||
|
Vascular disorders |
Hot flushes. |
Epistaxis, bleeding under skin, hypertension, high blood pressure aggravated, flushing, low blood pressure. | |
|
Ear and labyrinthine disorders |
Ear pain. | ||
|
Eye disorders |
Cataracts, ocular abnormalities, visual impairment, dry eyes. | ||
|
Gastrointestinal |
Abdominal pain |
Peritonitis, nausea, |
|
disorders |
upper, vomiting, obstipation, flatulence, diarrhoea, abdominal distension. |
dry mouth, dyspepsia, frequent bowel movements, faecal incontinence, rectal haemorrhage, haemorrhoids, melaena, gingivitis, toothache, diverticulitis. | |
|
General disorders and administration site conditions |
Asthenia. |
Injection site pain, oedema peripheral, chest pain |
Injection site haematoma, injection site inflammation, pain, malaise, pyrexia, oedema. |
|
Immune system disorders |
Allergic reactions, itching. |
Urticaria. | |
|
Infections and Infestations |
Shingles, urinary tract infection. | ||
|
Injury, poisoning and investigational complications |
Injury-non-site specific injuries NEC. | ||
|
Investigations |
Hepatic enzyme increased- liver enzyme increased, weight gain. |
Blood phosphokinase creatine increased, weight loss, cardiac murmur, breath sounds abnormal. | |
|
Metabolic and nutrition disorders |
Increased appetite, lipid metabolism disorder, hyperlipidemia, hypertriglyceridaemia, worsening of diabetes, gout. | ||
|
Musculoskeletal and connective tissue disorders |
Muscle weakness, myalgia, leg pain. |
Back pain, joint pain, arthritis aggravated, bursitis, muscle atrophy. | |
|
Nervous system disorders |
Dizziness, headaches, paraesthesia. |
Ataxia, coordination disturbances, speech disorders, cramp in legs, gait disturbance, abnormal touch sensation, migraine aggravated, |
|
facial pain (trigeminal neuralgia), syncope. | |||
|
Psychiatric disorders |
Anorexia nervosa, depression, sleep disturbances, somnolence. |
Agitation, anxiety, memory loss, concentration loss, confusion, depression aggravated, abnormal dreams, affect lability, nervousness, neurosis. | |
|
Renal and urinary disorders |
Pollakuria, nocturia, strangury. |
Haematuria, micturition disorder, urinary incontinence, urinary retention, urine flow decreased, urinary abnormality, dysuria. | |
|
Reproductive system and breast disorders |
Gynaecomastia. |
Nipple pain, impotence, loss of libido, testicular pain, testicular disorders. |
Epididymitis, penis disorder, prostatic disorder. |
|
Respiratory system, thoracic and mediastinal disorders |
Breathlessness. |
Bronchitis, coughing, pharyngeal mucositis (pharyngeal catarrh), pulmonary oedema, nasopharyngeal disorder (nasal mucositis), sinusitis, upper respiratory tract inflammation. | |
|
Skin and subcutaneous tissue disorders |
Alopecia (Hair loss), rash. |
Acne, rash-vesicular-vesicular eruption, dermatitis, erythema, dry skin, genital pruritus, genital itching, hair disorder, hyperhidrosis, chills, skin nodules, skin reaction. |
Comparison of treatment-induced undesirable effects up to Day 169 in three major clinical trials and two further trials shows that there is a similar incidence of all these undesirable effects. There were some noteworthy differences, however.
• The occurrence of back pain in the major clinical trials was 4% and 3% respectively, compared with >10% in the secondary studies. This may be attributable to the cumulative occurrence of metastatic diseases of the skeletal system in the patient group in the secondary studies.
• Obstipation and diarrhoea were more frequent in one of the secondary studies, in which patients with advanced symptomatic disease were represented, than in the major trials (obstipation: 5% compared with 1%; diarrhoea 5% compared with 3%). These differences may be attributable in certain circumstances to greater use of narcoanalgesics in patients with advanced symptomatic diseases, including painful skeletal metastases.
• In a clinical trial with an active comparative medicinal preparation, which compared Plenaxis with a combination of an LHRH agonist and a non-steroid anti-androgen, periodic ECGs were carried out. In both treatment groups, the QT interval, corrected in accordance with Fridericia’s formula, was prolonged by more than 10 milliseconds starting out from the baseline. In 20% of patients in both groups, there were either deviations by >30 milliseconds from the basic QTc interval, or QTc values at the end of treatment amounting to more than 450 milliseconds. Similar results were obtained in two other Phase III trials, in which Plenaxis was compared with an active treatment principle. It is unclear whether these changes were directly related to the test medicinal preparations, the androgen withdrawal or other variables.
Treatment with Plenaxis over a long period can lead to reduced bone mineral density.
A reduction in glucose tolerance has been observed in men who have had orchiectomy
or who have been treated with a GnRH agonist.
4.9 Overdose
No case of overdose has been reported. In clinical trials, 6 patients with prostate cancer received dosages of up to 150 mg (total quantity, 450 mg in the first 4 weeks). This dose did not have any undesirable effects differing in any way from those observed at a dosage of 100 mg of Plenaxis.
There is no specific antidote. Treatment must be symptom-related and supportive.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other hormone antagonists and related agents, ATC code: L02BX01
Mechanism of action
Plenaxis is a synthetic decapeptide, which functions as a gonadotropin-releasing hormone antagonist (GnRH antagonist). It causes a rapid reduction in serum levels of luteinising hormone (LH) and serum levels of follicle-stimulating hormone (FSH) and, as a consequence, the testosterone level in men and the oestradiol level in women.
This effect is reversible when treatment has been completed.
After the first injection of 100 mg, the testosterone level falls to castration level (<50 ng/dl) by the end of the first week in 70% of Plenaxis patients. Following a second injection on day 15, 94% of patients are castrated after 4 weeks. As Plenaxis is a GnRH antagonist, testosterone is lowered without prior flare. The results from studies 149-98-02 and 149-98-03 are typical of the differences observed between initiation with Plenaxis and a GnRH agonist and are discussed further below. At Day 30 the same testosterone level is reached following the different treatment options (Plenaxis, GnRH agonist or GnRH plus non-steroidal anti-androgen). Testosterone escape was more common in the Plenaxis arm of the comparative studies post day 85.
The effectiveness of Plenaxis in suppressing serum testosterone was studied in two randomised, open-label, active comparator trials (Studies 149-98-02 and 149-98-03). Patients were randomised in a 2:1 ratio to Plenaxis 100 mg IM versus GnRH agonist (Study 149-98-02) or to Plenaxis versus GnRH agonist with a non-steroidal antiandrogen (Study 149-98-03). Plenaxis was administered intramuscularly on Days 1, 15, 29 (Week 4), then every 4 weeks thereafter. The GnRH agonist and nonsteroidal anti-androgen were administered in the standard fashion.
In both studies combined, 100% (348/348) of Plenaxis patients and 16% (28/172) of comparator patients avoided a testosterone surge.
Serum testosterone levels as measured in studies 149-98-02 and 149-98-03 are given below (Figure 1 and Figure 2).
Figure 1: Serum testosterone levels measured in patients from study 149-98-02
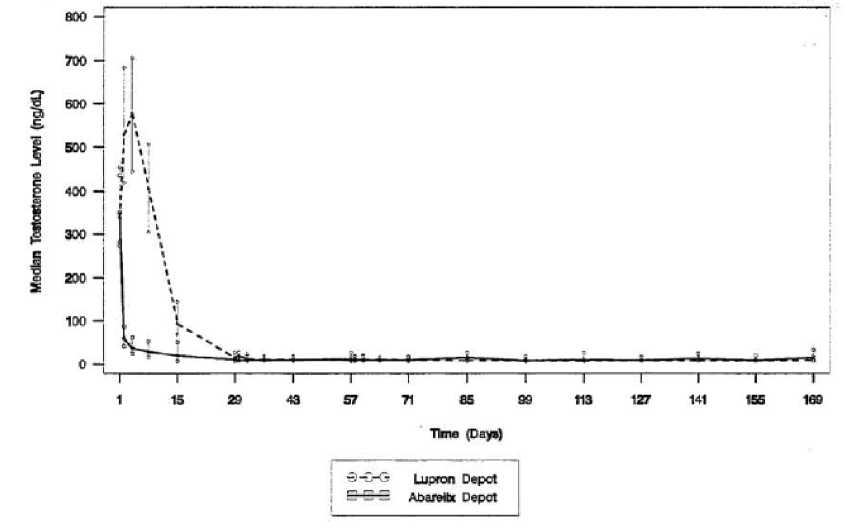
Figure 2: Serum testosterone levels measured in patients from study 149-98-03
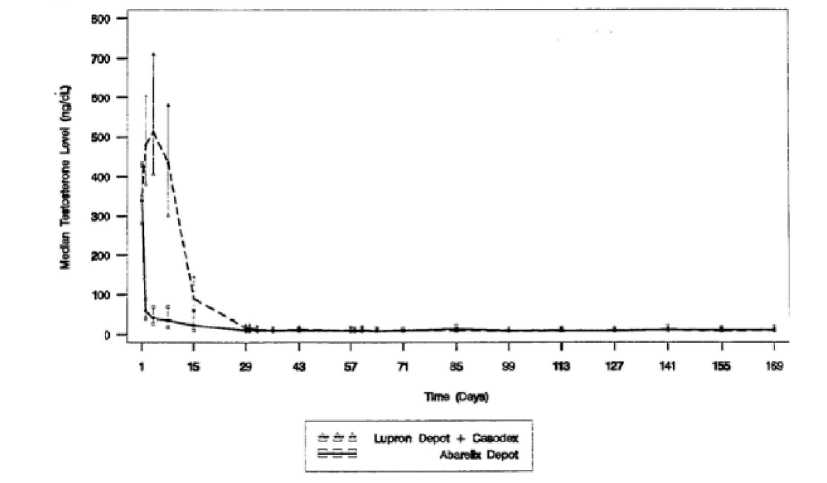
A successful response was defined as attainment of medical castration on Day 29 and maintenance through Day 85 (where no two consecutive serum testosterone concentrations between Days 29 and 85 were greater than 50 ng/dL). In Study 14998-02, 92% of Plenaxis patients responded and 96% of GnRH agonist patients responded. In Study 149-98-03, 93% of Plenaxis patients and 95% of GnRH agonist + non-steroidal antiandrogen patients responded. However, when failure was defined as any observed serum testosterone > 50 ng/dL (including transient elevations) just prior to dosing on Day 29 and every 28 days thereafter, effectiveness of testosterone suppression decreased over time.
The European Medicines Agency has waived the obligation to submit the results of studies with Plenaxis in all subsets of the paediatric population in prostate carcinoma (see section 4.2 for information on paediatric use).
5.2 Pharmacokinetic properties
A single dose of Plenaxis (100 mg intramuscularly) was administered to 14 healthy trial participants aged from 52 to 75 years and with a body weight ranging from 61.6 to 110.5 kg.
Resorption
Following the intramuscular administration of 100 mg of Plenaxis, abarelix is resorbed slowly. A mean peak concentration of 43.4 (48)ng/ml was measured some 3 days after the injection. The exposure of abarelix, in terms of AUC0-<x>, is approximately 500 ng/mL/day.
Body weight appeared to be a statistically significant covariate to the abarelix concentrations in both the pivotal clinical studies, although during the first 2 months of initiation of therapy. In Study 149-98-02, weight appeared to be a significant covariate on study days 2, 15, 29, 30, 57 and 58 only. In Study 14998-03, the significant of weight as a covariate was observed only on days 2, 15, 30 and 58. There was a negative relationship whereby heavier patients had lower abarelix concentrations. As has been observed with many drugs, heavier people tend to have higher volume of distribution for the drug and hence, lower levels.
In Study 149-98-02, patients that failed to meet the castration criteria (efficacy), were of significantly higher body weight (mean 97.2 ± 18.3 versus 85.8 ± 16.2 kg; p<0.05, one-way ANOVA) than those who passed the castration criteria. The same difference was not observed in the two groups in Study 149-95-03.
Distribution
In-vitro binding to human plasma protein ranges from 96% to 99%.
Metabolism
In-vitro studies with hepatocytes (rat, ape and human) and in-vivo studies (in rats and apes) showed that the main metabolites of abarelix are formed via the hydrolysis of peptide bonds. There was no evidence of any significant quantity of oxidative or conjugated metabolites. There is no indication of the distribution of cytochrome P-450 in the metabolism of Plenaxis.
Excretion
In humans, approximately 13% of unchanged abarelix was found in the urine following an intramuscular injection of 15 pg/kg of abarelix solution. There was no evidence of metabolites in the urine. Renal clearance was 14.4 l/day or 10 ml/min.
5.3 Preclinical safety data
The treatment-induced effects in male and female mice and rats were very similar to those observed in castrated and ovariectomised control animals, and were compatible with the pharmacological effects of hormonal ablation.
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction. Abarelix is embryolethal in rats and rabbits. It cannot be excluded that abarelix is also teratogenic. Most of the malformations occurred in the abarelix-treated groups but data were inconclusive as there were too few litters and foetuses available for adequate evaluation. No carcinogenic potential was observed at dosages corresponding to up to 11 times the human dosage (rats) and 17 times the human dosage (mice) in mg/m2 (human equivalence dosage).
Plenaxis has very weak histamine-releasing potential, as could be seen from the total histamine release from rat peritoneal mast cells.
Environmental Risk Assessment (ERA)
Abarelix is intended for use in the clinic. If used at home or at other locations, waste should be returned to the clinic for final disposal.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Powder: Carmellose sodium.
Solvent: sodium chloride 9 mg/mL (0.9%) solution for injection.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf life
Unopened vials of Plenaxis: 4 years.
Unopened vials of Solvent: 3 years.
Reconstituted vials: After reconstitution, the product should be used immediately from a microbiological viewpoint. However, chemical and physical stability for up to 8 hours after reconstitution at room temperature (below 25°C) has been shown.
6.4 Special precautions for storage
Unopened vials: The medicinal product does not require any special storage conditions.
For storage conditions of the reconstituted medicinal product, see section 6.3.
6.5 Nature and contents of container
Abarelix 100 mg powder for injection for suspension: Tinted borosilicate glass vial (Type I), sealed with a siliconised chlorobutyl rubber stopper and hinged plastic lid.
Sodium chloride 9 mg/mL (0.9%) solution for injection: Clear glass ampoule. The ampoule contains 3 ml, of which only 2.2 ml is used for reconstitution.
Plenaxis is supplied as a kit, which includes in addition a syringe for reconstitution and a needle for administration.
Package sizes:
Kit for 1 injection
Kit for 3 injections
Not all pack sizes may be marketed.
6.6 Special precautions for disposal
Important! It is essential to follow these instructions fully to ensure that the full therapeutic dose is administered.
1. Use aseptic technique for all steps.
2. Plenaxis powder may have aggregated during storage. To ensure that the patient receives the full dose (100 mg), it is critical that Plenaxis powder be as fine as possible prior to addition of solvent. Hold the vial at an angle of 45° and tap repeatedly on a hard surface or shake vigorously to fully disperse the powder.
Important! Look for clumps of powder by inspecting the vial whilst rolling it back and forth on its side. Do not proceed to the next stage without ensuring that all the lumps have been removed and the contents are in the form of a fine powder.
3. Withdraw 2.2 ml of the solvent (0.9% sodium chloride for injection purposes) using the enclosed syringe and 18G needle. Discard the remaining solvent.
4. Important! Keeping the vial upright (base down), insert the needle all the way into the vial until it cannot be inserted any further. Position the tip of the needle over the dimple at the base of the vial. Inject the solvent vigorously to ensure maximum dispersion of powder.
5. Before withdrawing the needle, remove 2.2 ml of air. Now remove the needle from the vial and expel the air.
6. Important! Do not shake the vial as this causes foaming. Foaming must be avoided to ensure that all the suspension is taken up into the syringe. Tip, roll and swirl the vial for 15 seconds and allow to stand for 2 minutes. During the 2 minutes, tap the vial and swirl it occasionally to reduce any foam.
7. Repeat step 6 one more time.
8. Insert the 18G needle back into the vial. Invert the vial and ‘wash’ the walls by withdrawing and gently reinjecting the suspension. Inspect the walls for any particulate matter. Repeat reinjection of suspension as many times as necessary to ensure all Plenaxis powder is completely in suspension.
9. Holding the vial at a 45° angle, withdraw the entire contents (at least 2 ml). Do not withdraw the needle until all the Plenaxis suspension has been drawn into the syringe. There should be no residual lumps or powder on the inner wall of the vial.
10. After withdrawing the needle, pull the syringe plunger back to draw up the residual suspension in the 18G needle into the syringe.
11. Now replace the 18G needle with the supplied 22G needle.
12. Insert the needle at the desired injection site and pull back the plunger to check for back-flow of blood. If blood flows into the syringe, do not inject at this site. Select another injection site.
13. Immediately administer the entire reconstituted suspension intramuscularly.
14. Observe the patient for approximately 30 minutes for any sign of an allergic or hypersensitive reaction.
Any unused product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
Speciality European Pharma Limited
14 Took’s Court
London
EC4A 1LB
United Kingdom
8 MARKETING AUTHORISATION NUMBER(S)
[To be completed nationally]
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE
AUTHORISATION
10 DATE OF REVISION OF THE TEXT
13/12/2013