Zoledronic Acid Ibigen 4 Mg/5 Ml Concentrate For Solution For Infusion
SUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Zoledronic Acid IBIGEN 4 mg/5 ml concentrate for solution for infusion
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
One vial with 5 ml concentrate contains 4 mg zoledronic acid (anhydrous), corresponding to 4.66 mg zoledronic acid hemipentahydrate.
One ml concentrate contains zoledronic acid (as hemipentahydrate) corresponding to 0.8 mg zoledronic acid (anhydrous).
For a full list of excipients, see section 6.1.
3 PHARMACEUTICAL FORM
Concentrate for solution for infusion. Clear and colourless solution.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
• Prevention of skeletal related events (pathological fractures, spinal compression, radiation or surgery to bone, or tumour-induced hypercalcaemia) in adult patients with advanced malignancies involving bone
• Treatment of adult patients with tumour-induced hypercalcaemia (TIH)
4.2 Posology and method of administration
Zoledronic Acid IBIGEN must only be used by clinicians experienced in the administration of intravenous bisphosphonates.
Zoledronic Acid IBIGEN reconstituted solution must not be mixed with calcium or other divalent cation-containing infusion solutions such as lactated Ringer’s solution, and should be administered as a single intravenous solution in a separate infusion line.
Prevention of skeletal related events in patients with advanced malignancies involving bone
Adults and elderly
The recommended dose in the prevention of skeletal related events in patients with advanced malignancies involving bone is 4 mg reconstituted and further diluted Zoledronic Acid IBIGEN solution for infusion (diluted with 100 ml 0.9% w/v sodium chloride or 5% w/v glucose solution), given in no less than a 15-minute intravenous infusion every 3 to 4 weeks. Patients should also be administered an oral calcium supplement of 500 mg and 400 IU vitamin D daily.
The decision to treat patients with bone metastases for the prevention of skeletal related events should consider that the onset of treatment effect is 2-3 months.
Treatment of TIH
Adults and elderly
The recommended dose in hypercalcaemia (albumin-corrected serum calcium > 12.0 mg/dl or 3.0 mmol/l) is 4 mg reconstituted and further diluted Zoledronic Acid IBIGEN solution for infusion (diluted with 100 ml sterile 0.9% w/v sodium chloride or 5% w/v glucose solution), given as a single intravenous infusion in no less than 15 minutes. Patients must be maintained well hydrated prior to and following administration of Zoledronic Acid IBIGEN.
Renal impairment
TIH:
Zoledronic Acid IBIGEN treatment in TIH patients who also have severe renal impairment should be considered only after evaluating the risks and benefits of treatment. In the clinical studies, patients with serum creatinine > 400 pmol/l or > 4.5 mg/dl were excluded. No dose adjustment is necessary in TIH patients with serum creatinine < 400 gmol/l or < 4.5 mg/dl (see section 4.4).
Prevention of skeletal related events in patients with advanced malignancies involving bone:
When initiating treatment with Zoledronic Acid IBIGEN in patients with multiple myeloma or metastatic bone lesions from solid tumours, serum creatinine and creatinine clearance (CLcr) should be determined. CLcr is calculated from serum creatinine using the Cockcroft-Gault formula. Zoledronic Acid IBIGEN is not recommended for patients presenting with severe renal impairment prior to initiation of therapy, which is defined for this population as CLcr < 30 ml/min. In clinical trials with Zoledronic Acid IBIGEN, patients with serum creatinine > 265 pmol/l or > 3.0 mg/dl were excluded. In patients with bone metastases presenting with mild to moderate renal impairment prior to initiation of therapy, which is defined for this population as CLcr 30-60 ml/min, the following Zoledronic Acid IBIGEN dose is recommended (see also section 4.4):
|
Baseline Creatinine Clearance (ml/min) |
Zoledronic Acid IBIGEN |
|
Recommended Dose* | |
|
> 60 |
4.0 mg |
|
50-60 |
3.5 mg* |
|
40-49 |
3.3 mg* |
|
30-39 |
3.0 mg* |
*Doses have been calculated assuming target AUC of 0.66 (mg*hr/l) (CLcr=75 ml/min). The reduced doses for patients with renal impairment are expected to achieve the same AUC as that seen in patients with creatinine clearance of 75 ml/min.
Following initiation of therapy, serum creatinine should be measured prior to each dose of Zoledronic Acid IBIGEN and treatment should be withheld if renal function has deteriorated. In the clinical trials, renal deterioration was defined as follows:
• For patients with normal baseline serum creatinine (< 1.4 mg/dl or < 124 pmol/l), an increase of 0.5 mg/dl or 44 pmol/l
• For patients with an abnormal baseline creatinine (> 1.4 mg/dl or > 124 pmol/l), an increase of 1.0 mg/dl or 88 pmol/l
In the clinical studies, zoledronic acid treatment was resumed only when the creatinine level returned to within 10% of the baseline value (see section 4.4). Zoledronic Acid IBIGEN treatment should be resumed at the same dose as that prior to treatment interruption.
Paediatric population
The safety and efficacy of zoledronic acid in children aged 1 year to 17 years have not been established. Currently available data are described in sections 4.4 and 5.1 but no recommendation on a posology can be made.
Method of administration
Intravenous use.
Zoledronic Acid IBIGEN 4 mg concentrate for solution for infusion, further diluted in 100 ml (see section 6.6), should be given as a single intravenous infusion in no less than 15 minutes.
In patients with mild to moderate renal impairment, reduced Zoledronic Acid IBIGEN doses are recommended (see section “Posology” above and section 6.3).
Instructions for preparing reduced doses of Zoledronic Acid IBIGEN
Withdraw an appropriate volume of the reconstituted solution (4 mg/5 ml) as needed:
• 4.4 ml for 3.5 mg dose
• 4.1 ml for 3.3 mg dose
• 3.8 ml for 3.0 mg dose
For information on the dilution of Zoledronic Acid IBIGEN, see section 6.6. The withdrawn amount of concentrate must be diluted in 100 ml of sterile 0.9% w/v sodium chloride solution or 5% w/v glucose solution. The dose must be given as a single intravenous infusion over no less than 15 minutes.
The use of Zoledronic Acid in paediatric patients has been studied in 2 clinical trials in the treatment of severe osteogenesis imperfecta (see section 5.1). Zoledronic Acid IBIGEN should not be used in the paediatric population because safety and efficacy in children have not been established (see sections 4.4 and 5.1).
Patients must be maintained well hydrated prior to and following administration of Zoledronic Acid IBIGEN.
4.3
Contraindications
• Hypersensitivity to the active substance, to other bisphosphonates or to any of the excipients in the formulation of Zoledronic Acid IBIGEN (see section 6.1)
• Breast-feeding (see section 4.6)
4.4 Special warnings and precautions for use
General
Patients must be assessed prior to administration of Zoledronic Acid IBIGEN to ensure that they are adequately hydrated. Overhydration should be avoided in patients at risk of cardiac failure.
Standard hypercalcaemia-related metabolic parameters, such as serum levels of calcium, phosphate and magnesium, should be carefully monitored after initiating Zoledronic Acid IBIGEN therapy. If hypocalcaemia, hypophosphataemia, or hypomagnesaemia occurs, short-term supplemental therapy may be necessary. Untreated hypercalcaemia patients generally have some degree of renal function impairment, therefore careful renal function monitoring should be considered.
Zoledronic Acid IBIGEN contains the same active substance as found in Aclasta (zoledronic acid). Patients being treated with Zoledronic Acid IBIGEN should not be treated with Aclasta or any other bisphosphonate concomitantly, since the combined effects of these agents are unknown.
The safety and efficacy of Zoledronic Acid in paediatric patients have not been established (see section 5.1).
Renal insufficiency
Patients with TIH with evidence of deterioration in renal function should be appropriately evaluated with consideration given as to whether the potential benefit of treatment with Zoledronic Acid IBIGEN outweighs the possible risk.
The decision to treat patients with bone metastases for the prevention of skeletal related events should consider that the onset of treatment effect is 2-3 months.
As with other bisphosphonates, Zoledronic Acid has been associated with reports of renal dysfunction. Factors that may increase the potential for deterioration in renal function include dehydration, pre-existing renal impairment, multiple cycles of Zoledronic Acid and other bisphosphonates as well as use of other nephrotoxic drugs. While the risk is reduced with a dose of Zoledronic Acid IBIGEN 4 mg administered over 15 minutes, deterioration in renal function may still occur. Renal deterioration, progression to renal failure and dialysis have been reported in patients after the initial dose or a single dose of Zoledronic Acid IBIGEN. Increases in serum creatinine also occur in some patients with chronic administration of Zoledronic Acid IBIGEN at recommended doses for prevention of skeletal related events, although less frequently.
Patients should have their serum creatinine levels assessed prior to each dose of Zoledronic Acid IBIGEN. Upon initiation of treatment in patients with bone metastases with mild to moderate renal impairment, lower doses of Zoledronic Acid IBIGEN are recommended. In patients who show evidence of renal deterioration during treatment, Zoledronic Acid IBIGEN should be withheld. Zoledronic Acid IBIGEN should only be resumed when serum creatinine returns to within 10% of baseline (see section 4.2). Zoledronic Acid IBIGEN treatment should be resumed at the same dose as that given prior to treatment interruption.
In view of the potential impact of bisphosphonates, including Zoledronic Acid, on renal function, the lack of clinical safety data in patients with severe renal impairment (in clinical trials defined as serum creatinine > 400 qmol/l or > 4.5 mg/dl for patients with TIH and > 265 qmol/l or > 3.0 mg/dl for patients with cancer and bone metastases, respectively) at baseline and only limited pharmacokinetic data in patients with severe renal impairment at baseline (creatinine clearance < 30 ml/min), the use of Zoledronic Acid IBIGEN is not recommended in patients with severe renal impairment.
Hepatic insufficiency
As only limited clinical data are available in patients with severe hepatic insufficiency, no specific recommendations can be given for this patient population.
Osteonecrosis of the jaw
Osteonecrosis of the jaw has been reported in patients, predominantly those with cancer, receiving treatment with bisphosphonates, including Zoledronic Acid. Many of these patients were also receiving chemotherapy and corticosteroids. The majority of reported cases have been associated with dental procedures such as tooth extraction. Many had signs of local infection including osteomyelitis.
A dental examination with appropriate preventive dentistry should be considered prior to treatment with bisphosphonates in patients with concomitant risk factors (e.g. cancer, chemotherapy, corticosteroids, poor oral hygiene).
While on treatment, these patients should avoid invasive dental procedures if possible. For patients who develop osteonecrosis of the jaw while on bisphosphonate therapy, dental surgery may exacerbate the condition. For patients requiring dental procedures, there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of osteonecrosis of the jaw. Clinical judgement of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment.
Atypical fractures of the femur
Atypical subtrochanteric and diaphyseal femoral fractures have been reported with bisphosphonate therapy, primarily in patients receiving long-term treatment for osteoporosis. These transverse or short oblique fractures can occur anywhere along the femur from just below the lesser trochanter to just above the supracondylar flare. These fractures occur after minimal or no trauma and some patients experience thigh or groin pain, often associated with imaging features of stress fractures, weeks to months before presenting with a completed femoral fracture. Fractures are often bilateral; therefore the contralateral femur should be examined in bisphosphonate-treated patients who have sustained a femoral shaft fracture. Poor healing of these fractures has also been reported. Discontinuation of bisphosphonate therapy in patients suspected to have an atypical femur fracture should be considered pending evaluation of the patient, based on an individual benefit risk assessment.
During bisphosphonate treatment patients should be advised to report any thigh, hip or groin pain and any patient presenting with such symptoms should be evaluated for an incomplete femur fracture.
Musculoskeletal pain
In post-marketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain have been reported in patients taking bisphosphonates. However, such reports have been infrequent. This category of drugs includes zoledronic acid. The time to onset of symptoms varied from one day to several months after starting treatment. Most patients had relief of symptoms after stopping treatment. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
4.5 Interaction with other medicinal products and other forms of interaction
In clinical studies, zoledronic acid has been administered concomitantly with commonly used anticancer agents, diuretics, antibiotics and analgesics without clinically apparent interactions occurring. Zoledronic acid shows no appreciable binding to plasma proteins and does not inhibit human P450 enzymes in vitro (see section 5.2), but no formal clinical interaction studies have been performed. Caution is advised when bisphosphonates are administered with aminoglycosides, since both agents may have an additive effect, resulting in a lower serum calcium level for longer periods than required. Caution is indicated when Zoledronic Acid IBIGEN is used with other potentially nephrotoxic drugs. Attention should also be paid to the possibility of hypomagnesaemia developing during treatment.
In multiple myeloma patients, the risk of renal dysfunction may be increased when intravenous bisphosphonates are used in combination with thalidomide.
4.6 Fertility, pregnancy and lactation
Pregnancy
There are no adequate data on the use of zoledronic acid in pregnant women. Animal reproduction studies with zoledronic acid have shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown. Zoledronic Acid IBIGEN should not be used during pregnancy.
Lactation
It is not known whether zoledronic acid is excreted into human milk. Zoledronic Acid IBIGEN is contraindicated in breast-feeding women (see section 4.3).
Fertility
Zoledronic acid was evaluated in rats for potential adverse effects on fertility of the parental and F1 generation. This resulted in exaggerated pharmacological effects considered related to the compounds inhibition of skeletal calcium metabolism, resulting in periparturient hypocalcaemia, a bisphosphonate class effect, dystocia and early termination of the study. Thus these results precluded determining a definitive effect of Zoledronic acid on fertility in humans.
4.7 Effects on ability to drive and use machines
Adverse reactions, such as dizziness and somnolence, may affect the ability to drive or use machines, therefore caution should be exercised with the use of Zoledronic Acid IBIGEN along with driving and operating of machinery.
4.8 Undesirable effects
Summary of the safety profile
Within three days after Zoledronic Acid administration, an acute phase reaction has commonly been reported, with symptoms including bone pain, fever, fatigue, arthralgia, myalgia and rigors; these symptoms usually resolve within a few days (see description of selected adverse reactions).
The following are the important identified risks with Zoledronic Acid IBIGEN in the approved indications: Renal function impairment, osteonecrosis of the jaw, acute phase reaction, hypocalcaemia, ocular adverse events, atrial fibrillation, anaphylaxis. The frequencies for each of these identified risks are shown in Table 1.
Tabulated list of adverse reactions
The following adverse reactions, listed in Table 1, have been accumulated from clinical studies and post-marketing reports following predominantly chronic treatment with 4 mg zoledronic acid:
Table 1
Adverse reactions are ranked under headings of frequency, the most frequent first, using the following convention: Very common (>1/10), common (>1/100 to <1/10), uncommon (>1/1,000 to <1/100), rare (>1/10,000 to <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data).
Blood and lymphatic system disorders
Common:
Uncommon:
Rare:
Immune system disorders Uncommon:
Rare:
Psychiatric disorders Uncommon:
Rare:
Nervous system disorders Common:
Uncommon:
Eye disorders
Common:
Uncommon:
Very rare:
Cardiac disorders Uncommon:
Anaemia
Thrombocytopenia, leukopenia Pancytopenia
Hypersensitivity reaction Angioneurotic oedema
Anxiety, sleep disturbance Confusion
Headache
Dizziness, paraesthesia, taste disturbance, hypoaesthesia, hyperaesthesia, tremor, somnolence
Conjunctivitis
Blurred vision, scleritis and orbital
inflammation
Uveitis, episcleritis
Hypertension, hypotension, atrial fibrillation, hypotension leading to
syncope or circulatory collapse
Rare: Bradycardia
Respiratory, thoracic and mediastinal disorders
Uncommon: Dyspnoea, cough, bronchoconstriction
Gastrointestinal disorders
Common: Nausea, vomiting, anorexia
Uncommon: Diarrhoea, constipation, abdominal pain,
dyspepsia, stomatitis, dry mouth Skin and subcutaneous tissue disorders
Uncommon: Pruritus, rash (including erythematous and
macular rash), increased sweating
Musculoskeletal and connective tissue disorders
Common:
Uncommon:
Renal and urinary disorders
Common:
Uncommon:
Bone pain, myalgia, arthralgia, generalised pain
Muscle cramps, osteonecrosis of the jaw* Renal impairment
Acute renal failure, haematuria, proteinuria
General disorders and administration site conditions
Common:
Uncommon:
Fever, flu-like syndrome (including fatigue, rigors, malaise and flushing) Asthenia, peripheral oedema, injection site reactions (including pain, irritation, swelling, induration), chest pain, weight increase, anaphylactic reaction/shock, urticaria
Investigations
Very common: Common:
Hypophosphataemia
Blood creatinine and blood urea increased, hypocalcaemia
Uncommon: Hypomagnesaemia, hypokalaemia
Rare: Hyperkalaemia, hypernatraemia
* Based on clinical trials with adjudication of possible cases of osteonecrosis of the jaw. Since these reports are subject to confounding factors, it is not possible to reliably establish a causal relationship to exposure to the medicinal product.
Description of selected adverse reactions
Renal function impairment
Zoledronic Acid has been associated with reports of renal dysfunction. Factors that may increase the potential for deterioration in renal function include dehydration, preexisting renal impairment, multiple cycles of Zoledronic Acid or other bisphosphonates, as well as concomitant use of nephrotoxic medicinal products or using a shorter infusion time than currently recommended. Renal deterioration, progression to renal failure and dialysis have been reported in patients after the initial dose or a single dose of 4 mg zoledronic acid (see section 4.4).
Osteonecrosis of the jaw
Cases of osteonecrosis (primarily of the jaws) have been reported, predominantly in cancer patients treated with medicinal products that inhibit bone resorption, such as Zoledronic Acid. Many of these patients had signs of local infection including osteomyelitis, and the majority of the reports refer to cancer patients following tooth extractions or other dental surgeries. Osteonecrosis of the jaws has multiple documented risk factors including a diagnosis of cancer, concomitant therapies (e.g. chemotherapy, radiotherapy, corticosteroids) and co-morbid conditions (e.g. anaemia, coagulopathies, infection, pre-existing oral disease). Although causality has not been determined, it is recommended to avoid dental surgery as recovery may be prolonged (see section 4.4).
Atrial fibrillation
In one 3-year, randomised, double-blind controlled trial that evaluated the efficacy and safety of zoledronic acid 5 mg once yearly vs. placebo in the treatment of postmenopausal osteoporosis (PMO), the overall incidence of atrial fibrillation was 2.5% (96 out of 3,862) and 1.9% (75 out of 3,852) in patients receiving zoledronic acid 5 mg and placebo, respectively. The rate of atrial fibrillation serious adverse events was 1.3% (51 out of 3,862) and 0.6% (22 out of 3,852) in patients receiving zoledronic acid 5 mg and placebo, respectively. The imbalance observed in this trial has not been observed in other trials with zoledronic acid, including those with Zoledronic Acid 4 mg every 3-4 weeks in oncology patients. The mechanism behind the increased incidence of atrial fibrillation in this single clinical trial is unknown.
Acute phase reaction
This adverse drug reaction consists of a constellation of symptoms that includes fever, myalgia, headache, extremity pain, nausea, vomiting, diarrhoea and arthralgia. The onset time is < 3 days post-Zoledronic Acid IBIGEN infusion, and the reaction is also referred to using the terms “flu-like” or “post-dose” symptoms.
Atypical fractures of the femur
During post-marketing experience the following reactions have been reported (frequency rare): Atypical subtrochanteric and diaphyseal femoral fractures (bisphosphonate class adverse reaction).
4.9 Overdose
Clinical experience with acute overdose of zoledronic acid is limited. Patients who have received doses higher than those recommended should be carefully monitored, since renal function impairment (including renal failure) and serum electrolyte (including calcium, phosphorus and magnesium) abnormalities have been observed.
In the event of hypocalcaemia, calcium gluconate infusions should be administered as clinically indicated.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Bisphosphonate, ATC code: M05 BA 08
Zoledronic acid belongs to the class of bisphosphonates and acts primarily on bone. It is an inhibitor of osteoclastic bone resorption.
The selective action of bisphosphonates on bone is based on their high affinity for mineralised bone, but the precise molecular mechanism leading to the inhibition of osteoclastic activity is still unclear. In long-term animal studies, zoledronic acid inhibits bone resorption without adversely affecting the formation, mineralisation or mechanical properties of bone.
In addition to being a potent inhibitor of bone resorption, zoledronic acid also possesses several antitumour properties that could contribute to its overall efficacy in the treatment of metastatic bone disease. The following properties have been demonstrated in preclinical studies:
- In vivo: Inhibition of osteoclastic bone resorption, which alters the bone marrow microenvironment, making it less conducive to tumour cell growth, anti-angiogenic activity and anti-pain activity.
- In vitro: Inhibition of osteoblast proliferation, direct cytostatic and pro-apoptotic activity on tumour cells, synergistic cytostatic effect with other anti-cancer drugs, anti-adhesion/invasion activity.
Clinical trial results in the prevention of skeletal related events in patients with advanced malignancies involving bone
The first randomised, double-blind, placebo-controlled study compared zoledronic acid to placebo for the prevention of skeletal related events (SREs) in prostate cancer patients. Zoledronic acid 4 mg significantly reduced the proportion of patients experiencing at least one skeletal related event (SRE), delayed the median time to first SRE by > 5 months, and reduced the annual incidence of events per patient -skeletal morbidity rate. Multiple event analysis showed a 36% risk reduction in developing SREs in the zoledronic acid group compared with placebo. Patients receiving zoledronic acid reported less increase in pain than those receiving placebo, and the difference reached significance at months 3, 9, 21 and 24. Fewer zoledronic acid patients suffered pathological fractures. The treatment effects were less pronounced in patients with blastic lesions. Efficacy results are provided in Table 2.
In a second study including solid tumours other than breast or prostate cancer, zoledronic acid 4 mg significantly reduced the proportion of patients with an SRE, delayed the median time to first SRE by > 2 months, and reduced the skeletal morbidity rate. Multiple event analysis showed 30.7% risk reduction in developing SREs in the zoledronic acid group compared with placebo. Efficacy results are provided in Table 3.
Table 2: Efficacy results (prostate cancer patients receiving hormonal therapy)
Radiation therapy
Any SRE (+TIH)
Fractures*
|
to bone | ||||||
|
Zoledroni c Acid 4 mg |
Placeb o |
Zoledroni c Acid 4 mg |
Placeb o |
Zoledroni c Acid 4 mg |
Placeb o | |
|
N |
214 |
208 |
214 |
208 |
214 |
208 |
|
Proportio n of patients with SREs (%) |
38 |
49 |
17 |
25 |
26 |
33 |
|
p-value |
0.028 |
0.052 |
0.119 | |||
|
Median time to SRE (days) |
488 |
321 |
NR |
NR |
NR |
640 |
|
p-value |
0.009 |
0.020 |
0.055 | |||
|
Skeletal morbidity rate |
0.77 |
1.47 |
0.20 |
0.45 |
0.42 |
0.89 |
|
p-value |
0.005 |
0.023 |
0.060 | |||
|
Risk reduction of suffering from multiple events** (%) |
36 |
NA |
NA |
NA |
NA | |
|
p-value |
0.002 |
NA |
NA | |||
* Includes vertebral and non-vertebral fractures
** Accounts for all skeletal events, the total number as well as time to each event during the trial
NR Not Reached
NA Not Applicable
Table 3: Efficacy results (solid tumours other than breast or prostate cancer)
|
Any SRE (+TIH) |
Fractures* |
Radiation therapy to bone | ||||
|
Zoledronic Acid 4 mg |
Placebo |
Zoledronic Acid 4 mg |
Placebo |
Zoledronic Acid 4 mg |
Placebo | |
|
N |
257 |
250 |
257 |
250 |
257 |
250 |
|
Proportion of patients with SREs (%) |
39 |
48 |
16 |
22 |
29 |
34 |
|
p-value |
0.039 |
0.064 |
0.173 | |||
|
Median time to SRE (days) |
236 |
155 |
NR |
NR |
424 |
307 |
|
p-value |
0.009 |
0.020 |
0.079 | |||
|
Skeletal morbidity rate |
1.74 |
2.71 |
0.39 |
0.63 |
1.24 |
1.89 |
|
p-value |
0.012 |
0.066 |
0.099 | |||
|
Risk reduction of suffering from multiple events** (%) |
30.7 |
NA |
NA |
NA |
NA | |
|
p-value |
0.003 |
NA |
NA | |||
* Includes vertebral and non-vertebral fractures
** Accounts for all skeletal events, the total number as well as time to each event during the trial
NR Not Reached
NA Not Applicable
In a third phase III randomised, double-blind trial, 4 mg zoledronic acid or 90 mg pamidronate every 3 to 4 weeks were compared in patients with multiple myeloma or breast cancer with at least one bone lesion. The results demonstrated that zoledronic acid 4 mg showed comparable efficacy to 90 mg pamidronate in the prevention of SREs. The multiple event analysis revealed a significant risk reduction of 16% in patients treated with zoledronic acid 4 mg in comparison with patients receiving pamidronate. Efficacy results are provided in Table 4.
Table 4: Efficacy results (breast cancer and multiple myeloma patients)
|
Any SRE (+TIH) |
Fractures* |
Radiation therapy to bone | ||||
|
Zoledronic Acid 4 mg |
Pam 90 mg |
Zoledronic Acid 4 mg |
Pam 90 mg |
Zoledronic Acid 4 mg |
Pam 90 mg | |
|
N |
561 |
555 |
561 |
555 |
561 |
555 |
|
Proportion of patients with SREs (%) |
48 |
52 |
37 |
39 |
19 |
24 |
|
p-value |
0.198 |
0.653 |
0.037 | |||
|
Median time to SRE (days) |
376 |
356 |
NR |
714 |
NR |
NR |
|
p-value |
0.151 |
0.672 |
0.026 | |||
|
Skeletal morbidity rate |
1.04 |
1.39 |
0.53 |
0.60 |
0.47 |
0.71 |
|
p-value |
0.084 |
0.614 |
0.015 | |||
|
Risk reduction of suffering from multiple events** (%) |
16 |
NA |
NA |
NA |
NA | |
|
p-value |
0.030 |
NA |
NA | |||
* Includes vertebral and non-vertebral fractures
** Accounts for all skeletal events, the total number as well as time to each event during the trial
NR Not Reached
NA Not Applicable
Zoledronic Acid was also studied in a double-blind, randomised, placebo-controlled trial in 228 patients with documented bone metastases from breast cancer to evaluate the effect of zoledronic acid on the skeletal related event (SRE) rate ratio, calculated as the total number of SRE events (excluding hypercalcaemia and adjusted for prior fracture), divided by the total risk period. Patients received either 4 mg zoledronic acid or placebo every four weeks for one year. Patients were evenly distributed between Zoledronic Acid -treated and placebo groups.
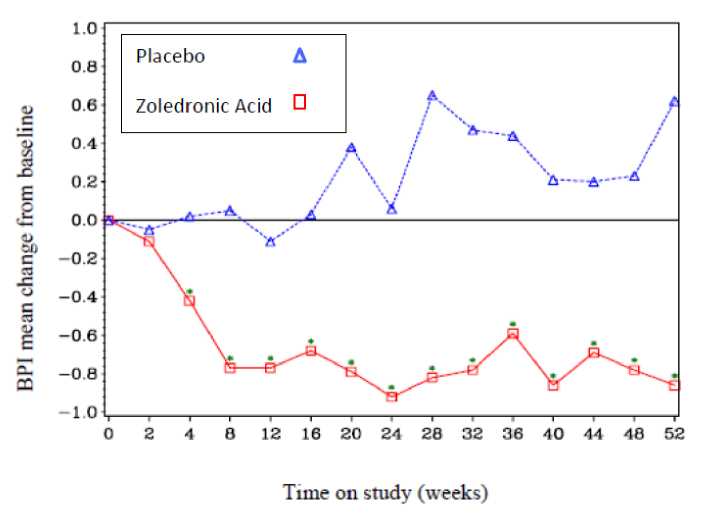
The SRE rate (events/person year) was 0.628 for zoledronic acid and 1.096 for placebo. The proportion of patients with at least one SRE (excluding hypercalcaemia) was 29.8% in the zoledronic acid-treated group versus 49.6% in the placebo group (p=0.003). Median time to onset of the first SRE was not reached in the zoledronic acid-treated arm at the end of the study and was significantly prolonged compared to placebo (p=0.007). Zoledronic acid reduced the risk of SREs by 41% in a multiple event analysis (risk ratio=0.59, p=0.019) compared with placebo.
In the Zoledronic Acid-treated group, statistically significant improvement in pain scores (using the Brief Pain Inventory, BPI) was seen at 4 weeks and at every subsequent time point during the study, when compared to placebo (Figure 1). The pain score for zoledronic acid was consistently below baseline and pain reduction was accompanied by a trend in reduced analgesics score.
Figure 1: Mean changes from baseline in BPI scores. Statistically significant differences are marked (*p<0.05) for between treatment comparisons (Zoledronic Acid vs. Placebo)
Clinical trial results in the treatment of TIH
Clinical studies in tumour-induced hypercalcaemia (TIH) demonstrated that the effect of zoledronic acid is characterised by decreases in serum calcium and urinary calcium excretion. In Phase I dose finding studies in patients with mild to moderate tumour-induced hypercalcaemia (TIH), effective doses tested were in the range of approximately 1.2-2.5 mg.
To assess the effects of zoledronic acid versus pamidronate 90 mg, the results of two pivotal multicentre studies in patients with TIH were combined in a pre-planned analysis. There was faster normalisation of corrected serum calcium at day 4 for zoledronic acid 8 mg and at day 7 for zoledronic acid 4 mg and 8 mg. The following response rates were observed:
Table 5: Proportion of complete responders by day in the combined TIH studies
Day 4
Day 7
Day 10
|
Zoledronic Acid 4 mg (N=86) |
45.3% (p=0.104) |
82.6% (p=0.005)* |
88.4% (p=0.002)* |
|
Zoledronic Acid 8 mg (N=90) |
55.6% (p=0.021)* |
83.3% (p=0.010)* |
86.7% (p=0.015)* |
|
Pamidronate 90 mg (N=99) |
33.3% |
63.6% |
69.7% |
|
*p-values compared to pamidronate | |||
Median time to normocalcaemia was 4 days. Median time to relapse (re-increase of albumin-corrected serum calcium > 2.9 mmol/l) was 30 to 40 days for patients treated with zoledronic acid versus 17 days for those treated with pamidronate 90 mg (p-values: 0.001 for 4 mg and 0.007 for 8 mg). There were no statistically significant differences between the two zoledronic acid doses.
In clinical trials 69 patients who relapsed or were refractory to initial treatment (zoledronic acid 4 mg, 8 mg or pamidronate 90 mg) were retreated with zoledronic acid 8 mg. The response rate in these patients was about 52%. Since those patients were retreated with the 8 mg dose only, there are no data available allowing comparison with the 4 mg dose.
In clinical trials performed in patients with tumour-induced hypercalcaemia (TIH), the overall safety profile amongst all three treatment groups (zoledronic acid 4 and 8 mg and pamidronate 90 mg) was similar in types and severity.
Paediatric population
Clinical trial results in the treatment of severe osteogenesis imperfecta in paediatric patients aged 1 to 17 years
The effects of intravenous zoledronic acid in the treatment of paediatric patients (age 1 to 17 years) with severe osteogenesis imperfecta (types I, III and IV) were compared to intravenous pamidronate in one international, multicentre, randomised, open-label study with 74 and 76 patients in each treatment group, respectively. The study treatment period was 12 months preceded by a 4- to 9-week screening period during which vitamin D and elemental calcium supplements were taken for at least 2 weeks. In the clinical programme patients aged 1 to < 3 years received 0.025 mg/kg zoledronic acid (up to a maximum single dose of 0.35 mg) every 3 months and patients aged 3 to 17 years received 0.05 mg/kg zoledronic acid (up to a maximum single dose of 0.83 mg) every 3 months. An extension study was conducted in order to examine the long-term general and renal safety of once yearly or twice yearly zoledronic acid over the 12-month extension treatment period in children who had completed one year of treatment with either zoledronic acid or pamidronate in the core study.
The primary endpoint of the study was the percent change from baseline in lumbar spine bone mineral density (BMD) after 12 months of treatment. Estimated treatment effects on BMD were similar, but the trial design was not sufficiently robust to establish non-inferior efficacy for zoledronic acid. In particular there was no clear evidence of efficacy on incidence of fracture or on pain. Fracture adverse events of long bones in the lower extremities were reported in approximately 24% (femur) and 14% (tibia) of zoledronic acid-treated patients vs 12% and 5% of pamidronate-treated patients with severe osteogenesis imperfecta, regardless of disease type and causality but overall incidence of fractures was comparable for the zoledronic acid and pamidronate-treated patients: 43% (32/74) vs 41% (31/76). Interpretation of the risk of fracture is confounded by the fact that fractures are common events in patients with severe osteogenesis imperfecta as part of the disease process.
The type of adverse reactions observed in this population were similar to those previously seen in adults with advanced malignancies involving the bone (see section 4.8). The adverse reactions ranked under headings of frequency, are presented in Table 6. The following conventional classification is used: very common (>1/10), common (>1/100, <1/10), uncommon (>1/1,000, <1/100), rare (>1/10,000, <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data).
Table 6: Adverse reactions observed in paediatric patients with severe osteogenesis imperfecta1
|
Nervous system disorders Common: |
Headache |
|
Cardiac disorders Common: |
Tachycardia |
|
Respiratory, thoracic and mediastinal disorders Common: |
Nasopharyngitis |
|
Gastrointestinal disorders Very common: Common: |
Vomiting, nausea Abdominal pain |
|
Musculoskeletal and connective tissue disorders Common: |
Pain in extremities, arthralgia, musculoskeletal pain |
|
General disorders and administration site conditions Very common : Common: |
Pyrexia, fatigue Acute phase reaction, pain |
|
Investigations Very common: Common: |
Hypocalcaemia Hypophosphataemia |
1 Adverse events occurring with frequencies < 5% were medically assessed and it was shown that these cases are consistent with the well-established safety profile of zoledronic acid (see section 4.8)
In paediatric patients with severe osteogenesis imperfecta, zoledronic acid seems to be associated with more pronounced risks for acute phase reaction, hypocalcaemia and unexplained tachycardia, in comparison to pamidronate, but this difference declined after subsequent infusions.
The European Medicines Agency has waived the obligation to submit the results of studies with zoledronic acid in all subsets of the paediatric population in the treatment of tumour-induced hypercalcaemia and prevention of skeletal-related events in patients with advanced malignancies involving bone (see section 4.2 for information on paediatric use).
5.2 Pharmacokinetic properties
Single and multiple 5- and 15-minute infusions of 2, 4, 8 and 16 mg zoledronic acid in 64 patients with bone metastases yielded the following pharmacokinetic data, which were found to be dose independent.
After initiating the infusion of zoledronic acid, the plasma concentrations of drug rapidly increased, achieving their peak at the end of the infusion period, followed by a rapid decline to < 10% of peak after 4 hours and < 1% of peak after 24 hours, with a subsequent prolonged period of very low concentrations not exceeding 0.1% of peak prior to the second infusion of drug on day 28.
Intravenously administered zoledronic acid is eliminated by a triphasic process: rapid biphasic disappearance from the systemic circulation, with half-lives of t'Aa 0.24 and t'AP 1.87 hours, followed by a long elimination phase with a terminal elimination half-life of t'Ay 146 hours. There was no accumulation of drug in plasma after multiple doses of the drug given every 28 days. Zoledronic acid is not metabolised and is excreted unchanged via the kidney. Over the first 24 hours, 39 ± 16% of the administered dose is recovered in the urine, while the remainder is principally bound to bone tissue. From the bone tissue it is released very slowly back into the systemic circulation and eliminated via the kidney. The total body clearance is 5.04 ± 2.5 l/h, independent of dose, and unaffected by gender, age, race, and body weight. Increasing the infusion time from 5 to 15 minutes caused a 30% decrease in zoledronic acid concentration at the end of the infusion, but had no effect on the area under the plasma concentration versus time curve.
The interpatient variability in pharmacokinetic parameters for zoledronic acid was high, as seen with other bisphosphonates.
No pharmacokinetic data for zoledronic acid are available in patients with hypercalcaemia or in patients with hepatic insufficiency. Zoledronic acid does not inhibit human P450 enzymes in vitro, shows no biotransformation and in animal studies < 3% of the administered dose was recovered in the faeces, suggesting no relevant role of liver function in the pharmacokinetics of zoledronic acid.
The renal clearance of zoledronic acid was correlated with creatinine clearance, renal clearance representing 75 ± 33% of the creatinine clearance, which showed a mean of 84 ± 29 ml/min (range 22 to 143 ml/min) in the 64 cancer patients studied. Population analysis showed that for a patient with creatinine clearance of 20 ml/min (severe renal impairment), or 50 ml/min (moderate impairment), the corresponding predicted clearance of zoledronic acid would be 37% or 72%, respectively, of that of a patient showing creatinine clearance of 84 ml/min. Only limited pharmacokinetic data are available in patients with severe renal insufficiency (creatinine clearance < 30 ml/min).
Zoledronic acid shows no affinity for the cellular components of blood and plasma protein binding is low (approximately 56%) and independent of the concentration of zoledronic acid.
Special populations
Paediatric patients
Limited pharmacokinetic data in children with severe osteogenesis imperfecta suggest that zoledronic acid pharmacokinetics in children aged 3 to 17 years are similar to those in adults at a similar mg/kg dose level. Age, body weight, gender and creatinine clearance appear to have no effect on zoledronic acid systemic exposure.
5.3 Preclinical safety data
Acute toxicity
The highest non-lethal single intravenous dose was 10 mg/kg bodyweight in mice and 0.6 mg/kg in rats.
Subchronic and chronic toxicity
Zoledronic acid was well tolerated when administered subcutaneously to rats and intravenously to dogs at doses up to 0.02 mg/kg daily for 4 weeks. Administration of 0.001 mg/kg/day subcutaneously in rats and 0.005 mg/kg intravenously once every 23 days in dogs for up to 52 weeks was also well tolerated.
The most frequent finding in repeat-dose studies consisted of increased primary spongiosa in the metaphyses of long bones in growing animals at nearly all doses, a finding that reflected the compounds pharmacological antiresorptive activity.
The safety margins relative to renal effects were narrow in the long-term repeat-dose parenteral animal studies but the cumulative no adverse event levels (NOAELs) in the single dose (1.6 mg/kg) and multiple dose studies of up to one month (0.06-0.6 mg/kg/day) did not indicate renal effects at doses equivalent to or exceeding the highest intended human therapeutic dose. Longer-term repeat administration at doses bracketing the highest intended human therapeutic dose of zoledronic acid produced toxicological effects in other organs, including the gastrointestinal tract, liver, spleen and lungs, and at intravenous injection sites.
Reproduction toxicity
Zoledronic acid was teratogenic in the rat at subcutaneous doses > 0.2 mg/kg. Although no teratogenicity or foetotoxicity was observed in the rabbit, maternal toxicity was found. Dystocia was observed at the lowest dose (0.01 mg/kg bodyweight) tested in the rat.
Mutagenicity and carcinogenic potential
Zoledronic acid was not mutagenic in the mutagenicity tests performed and carcinogenicity testing did not provide any evidence of carcinogenic potential.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Mannitol
Sodium citrate Water for injections
6.2 Incompatibilities
To avoid potential incompatibilities, Zoledronic Acid IBIGEN concentrate is to be diluted with 0.9% w/v sodium chloride solution or 5% w/v glucose solution.
Zoledronic Acid IBIGEN concentrate must not be mixed with calcium or other divalent cation-containing infusion solutions such as lactated Ringer’s solution, and should be administered as a single intravenous solution in a separate infusion line.
Studies with glass bottles, as well as several types of infusion bags and infusion lines made from polyvinylchloride, polyethylene and polypropylene (prefilled with 0.9% w/v sodium chloride solution or 5% w/v glucose solution), showed no incompatibility with Zoledronic Acid IBIGEN.
6.3 Shelf life
18 months.
From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2 to 8°C, unless reconstitution/dilution has taken place in controlled and validated aseptic conditions.
6.4 Special precautions for storage
No special precautions for storage.
For storage conditions of the reconstituted solution for infusion, see section 6.3.
6.5 Nature and contents of container
Zoledronic Acid IBIGEN 4 mg/5 ml concentrate for solution for infusion is supplied as pack containing 1 vial.
Vial: 5-ml plastic vial made of clear, colourless cycloolefine copolymer with fluoropolymer-coated bromobutyl rubber stopper and aluminium cap with plastic flip-off component.
6.6 Special precautions for disposal
Prior to administration, 5.0 ml concentrate from one vial or the volume of the concentrate withdrawn as required must be further diluted with 100 ml of calcium-free infusion solution (0.9% w/v sodium chloride solution or 5% w/v glucose solution). If refrigerated, the solution must be allowed to reach room temperature before administration.
Aseptic techniques must be followed during the preparation of the infusion. For single use only.
Only clear solution free from particles and discolouration should be used.
Healthcare professionals are advised not to dispose of unused Zoledronic Acid IBIGEN via the domestic sewage system.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
Ibigen S.r.l Via Fossignano, 2 04011 Aprilia (LT)
Italy
8 MARKETING AUTHORISATION NUMBER(S)
PL 31745/0017
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
22/02/2013
10 DATE OF REVISION OF THE TEXT
22/02/2013