Willfact 2000 I.U Powder And Solvent For Solution For Injection
Out of date information, search anotherSUMMARY OF PRODUCT CHARACTERISTICS
1 NAME OF THE MEDICINAL PRODUCT
Willfact 2000 IU
Powder and solvent for solution for injection.
2 QUALITATIVE AND QUANTITATIVE COMPOSITION
Willfact is presented as powder and solvent for solution for injection containing nominally 2000 IU human von Willebrand factor per vial.
Every 1 ml of reconstituted solution contains Willfact 100 IU human von Willebrand factor when reconstituted with 20 ml of water for injection.
The product contains approximately 100 IU/ml human von Willebrand factor when reconstituted with 20 ml of water for injections. Before the addition of albumin, the specific activity of Willfact 2000 IU is > 50 IU VWF:RCo/mg protein.
The von Willebrand factor potency (IU) is measured according to ristocetin cofactor activity (VWF:RCo) compared to the International Standard for von Willebrand factor concentrate.
The quantity of human factor VIII in Willfact is < 10 IU/100 IU VWF:RCo. The FVIII potency (IU) is determined using the European Pharmacopoeia chromogenic assay.
For a full list of excipients, see section 6.1. One vial (2000 IU) of Willfact contains 0.6 mmol (13.8 mg) sodium.
3 PHARMACEUTICAL FORM
Powder and solvent for solution for injection.
4 CLINICAL PARTICULARS
4.1 Therapeutic indications
Willfact is indicated in the prevention and treatment of haemorrhages or surgical bleeding in von Willebrand disease when desmopressin (DDAVP) treatment alone is ineffective or contraindicated.
Willfact should not be used in the treatment of haemophilia A.
Treatment of von Willebrand disease should be supervised by a physician experienced in the treatment of haemostatic disorders.
Generally, 1 IU/kg of von Willebrand factor raises the circulating level of VWF:RCo by 0.02 IU/ml (2 %).
Levels of VWF:RCo of > 0.6 IU/ml (60 %) and of FVIII:C of > 0.4 IU/ml (40 %) should be achieved.
Haemostasis cannot be ensured until FVIII coagulant activity (FVIII:C) has reached 0.4 IU/ml (40 %). A single injection of von Willebrand factor will only lead to a maximum increase in FVIII:C after at least 6-12 hours. The single administration of von Willebrand factor cannot immediately correct the FVIII:C level. If the patient’s baseline plasma FVIII:C level is below this critical level, in all situations where a rapid correction of haemostasis should be achieved, such as the treatment of haemorrhage, severe trauma or emergency surgery, it is necessary to administer a factor VIII product with the first injection of von Willebrand factor, in order to achieve a haemostatic plasma level of FVIII:C.
However, if an immediate rise in FVIII:C is not necessary, for example in a planned operation, or if the baseline FVIII:C level is sufficient to ensure haemostasis, the physician may decide to omit the co-administration of FVIII at the first injection.
Start of treatment
The first dose of Willfact is 40 to 80 IU/kg for the treatment of haemorrhage or trauma, in conjunction with the required amount of factor VIII product, calculated according to the patient's baseline plasma level of FVIII:C, in order to achieve an appropriate plasma level of FVIII:C, immediately before the intervention or as soon as possible after the onset of the bleeding episode or severe trauma. In case of surgery, it should be given 1 hour before the procedure.
An initial dose of 80 IU/kg of Willfact may be required, especially in patients with Type 3 von Willebrand disease where maintenance of adequate levels may require higher doses than in other types of VWD.
For elective surgery, treatment with Willfact should start 12-24 hours before surgery and should be repeated 1 hour before the procedure. In this case, co-administration of factor VIII product is not required since endogenous FVIII:C has usually reached the critical level of 0.4 IU/ml (40 %) before surgery. However, this should be confirmed in each patient.
Subsequent injections
If required, treatment should be continued with an appropriate dose of Willfact, with 40 - 80 IU/kg per day in 1 or 2 injections daily over one to several days. The dose and duration of the treatment depend on the clinical status of the patient, the type and severity of bleeding and both VWF:RCo and FVIII:C levels.
Long-term prophylaxis
Willfact can be administered as long-term prophylaxis in a dose which is determined individually for each patient. Willfact doses between 40 and 60 IU/kg, administered two to three times per week, reduce the number of haemorrhagic episodes.
There is no data from a clinical study to characterise the response to use of Willfact in children less than 6 years of age.
The use of Willfact in children under 12 years of age is only documented in individual cases; the use of Willfact in patients previously untreated with von Willebrand factor is not documented in the clinical studies.
Method of administration
Dissolve the preparation as described under 6.6. The product should be administered via the intravenous route at a maximum rate of 4 ml/minute.
4.3 Contraindications
Hypersensitivity to the active substance or any of the ingredients.
4.4 Special warnings and precautions for use
In actively bleeding patients it is recommended to co-administer a FVIII product with the von Willebrand factor product with a low FVIII content as a first line treatment.
As with any intravenous administration of a plasma-derived protein, allergy type hypersensitivity reactions are possible. Patients must be closely monitored and carefully observed for any symptoms throughout the injection period. Patients should be informed of the early signs of hypersensitivity reactions such as hives, generalised urticaria, tightness of the chest, wheezing, hypotension and anaphylaxis. If these symptoms occur, administration should be discontinued immediately. In case of shock, standard medical treatment for shock should be implemented.
Standard measures to prevent infections resulting from the use of medicinal products prepared from human blood or plasma include selection of donors, screening of individual donations and plasma pools for specific markers of infection and the inclusion of effective manufacturing steps for the inactivation/removal of viruses.
Despite this, when medicinal products prepared from human blood or plasma are administered, the possibility of transmitting infective agents cannot be totally excluded. This also applies to unknown or emerging viruses and other pathogens.
The measures taken are considered effective for enveloped viruses such as HIV, HBV and HCV. The measures taken may be of limited value against non-enveloped viruses such as HAV and parvovirus B19. Parvovirus B19 infection may be serious for pregnant women (foetal infection) and for individuals with immunodeficiency or increased erythropoesis (e.g. haemolytic anaemia).
Appropriate vaccination (hepatitis A and hepatitis B) should be considered for patients regularly receiving human plasma-derived von Willebrand factor.
Note that the name and batch number of the drug must be documented for every administration of Willfact to help establish a link between the patient and the batch number.
There is a risk of occurrence of thrombotic events, particularly in patients with known clinical or laboratory risk factors. Therefore, patients at risk must be monitored for early signs of thrombosis. Prophylaxis against venous thromboembolisms should be instituted according to the current recommendations.
When using a FVIII-containing VWF preparation, the treating physician should be aware that continued treatment may cause an excessive rise in FVIII:C. In patients receiving factor VIII-containing von Willebrand factor products, plasma levels of FVIII:C should be monitored to avoid sustained excessive FVIII:C plasma levels, which may increase the risk of thrombotic events.
Patients with von Willebrand disease, especially Type 3 patients, may develop neutralising antibodies (inhibitors) to VWF. If the expected VWF:RCo activity plasma levels are not attained, or if the bleeding cannot be controlled with an appropriate dose, an assay should be performed to determine if a VWF inhibitor is present. In patients with high levels of inhibitor, von Willebrand factor therapy may not be effective and other therapeutic options should be considered.
One vial (2000 IU) of Willfact contains 0.6 mmol (13.8 mg) sodium. For more than 3300 IU injected (more than 1 mmol sodium), to be taken into consideration by patients on a controlled sodium diet.
4.5 Interaction with other medicinal products and other forms of interaction
No interactions of human von Willebrand factor products with other medicinal products are known.
4.6 Fertility, pregnancy and lactation
Animal studies are insufficient to assess its safety with respect to fertility, reproduction, pregnancy, embryonic/fmtal development or peri- and postnatal development.
The safety of Willfact during pregnancy and lactation has not been investigated in controlled clinical studies.
Willfact should be administered to pregnant and lactating von Willebrand factor deficient women only if clearly indicated.
4.7 Effects on ability to drive and use machines
No effects on the ability to drive or use machines have been observed.
4.8 Undesirable effects
Although Willfact is considered safe, allergic or anaphylactic reactions may occur.
The undesirable effects listed below were assessed on the basis of the following frequency specifications:
Very common (>1/10)
Common (>1/100 to <1/10)
Uncommon (>1/1,000 to <1/100)
Rare (>1/10,000 to <1/1,000)
Very rare (<1/10,000),
not known (frequency cannot be estimated from the available data)
Immune system disorders
Uncommon: Hypersensitivity or allergic reactions. These may in some cases progress to severe anaphylaxis (including shock).
Psychiatric disorders
Uncommon: Restlessness
Nervous system disorders
Uncommon: Headache, tingling, lethargy
Cardiac disorders
Uncommon: Tachycardia
Vascular disorders
Uncommon: Hypotension, flushing
Respiratory thoracic and mediastinal disorders
Uncommon: Wheezing
Gastrointestinal disorders
Uncommon: Nausea, vomiting
Skin and subcutaneous tissue disorders
Uncommon: Angioedema, generalised urticaria, hives
General disorders and administration site conditions
Uncommon: Burning and stinging at the infusion site, chills, tightness of the chest
Rare: Fever
Investigations
Very rare: Neutralising antibodies (inhibitors) to VWF
Patients with von Willebrand disease, especially type 3 patients, may very rarely develop neutralising antibodies (inhibitors) to VWF. Patients treated with VWF should be carefully monitored for the development of inhibitors using appropriate clinical observations and laboratory tests. If such inhibitors occur, the condition will manifest itself as an inadequate clinical response. Such antibodies are precipitating and occur in close association with anaphylactic reactions. In all such cases, it is recommended that a specialised haemophilia centre be contacted.
Therefore, patients experiencing anaphylactic reaction should be evaluated for the presence of an inhibitor.
After correction of the factor Willebrand deficiency, due to the risk of a thrombotic episode in certain risk situations, monitoring for early signs of thrombosis or disseminated intravascular coagulation and prevention of thromboembolic complications should be undertaken according to current practices.
In patients receiving FVIII-containing VWF products sustained excessive FVIII:C plasma levels may increase the risk of thrombotic events.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via: "Yellow Card Scheme, Website: www.mhra.gov.uk/yellowcard".
4.9 Overdose
No case of overdose with Willfact has been reported. Thromboembolic events may occur in case of major overdose.
5 PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Anti-haemorrhagics: von Willebrand factor ATC code: B02BD10
Willfact behaves in the same way as endogenous von Willebrand factor.
Administration of von Willebrand factor allows correction of the haemostatic abnormalities exhibited by patients who suffer from von Willebrand factor deficiency at two levels:
• VWF re-establishes platelet adhesion to the vascular subendothelium at the site of vascular damage (as it binds both to the vascular subendothelium and to the platelet membrane) providing primary haemostasis as shown by the shortening of the bleeding time. This effect is known to depend to a large extent on the level of multimerisation of the active substance.
• Von Willebrand factor produces delayed correction of the associated factor VIII deficiency. Administered intravenously, von Willebrand factor binds to endogenous factor VIII (which is produced normally by the patient), and by stabilising this factor, avoids its rapid degradation. Because of this, administration of pure von Willebrand factor (vWF product with a low FVIII level) restores the FVIII:C level to normal as a secondary effect after the first
infusion. Administration of a FVIII:C containing VWF preparation restores the FVIII:C level to normal immediately after the first infusion.
5.2 Pharmacokinetic properties
A pharmacokinetic study with Willfact was carried out on 8 patients with type 3 von Willebrand disease. It demonstrated that for VWF:RCo:
• The mean AUC0-oo is 3444 IU.h/dl after single dose of 100 IU/kg Willfact,
• The plasma peak is reached between 30 minutes and 1 hour after injection,
• The mean recovery is 2.1 [IU/dl]/[IU/kg] of the injected preparation,
• The half-life is between 8 and 14 hours, with a mean value of 12 hours,
• The mean clearance is 3.0 ml/h/kg.
Normalisation of FVIII level is progressive, varies and usually requires between 6 and 12 hours. This effect is sustained for 2 to 3 days.
The increase in FVIII level is progressive and returns to normal after 6 to 12 hours. The FVIII level increases by a mean of 6 % (IU/dl) per hour. Thus, even in patients with an initial FVIII:C level less than 5 % (IU/dl), the FVIII:C level increases to around 40 % (IU/dl) 6 hours after the injection, and this level is maintained over 24 hours.
5.3 Preclinical safety data
Based on data obtained from several preclinical studies using animal models, there is no evidence for other toxic effect of Willfact than those related to the immunogenicity of human proteins in laboratory animals. Repeated dose toxicity testing is impracticable due to the development of antibodies to heterologous protein in animal models.
The preclinical safety data do not suggest that Willfact has any mutagenic potential.
6 PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Powder: human albumin, arginine hydrochloride, glycine, sodium citrate and calcium chloride dihydrate.
Solvent: water for injections.
6.2 Incompatibilities
Willfact must not be mixed with other medicinal products except for plasma-derived coagulation FVIII produced by LFB-BIOMEDICAMENTS, with which a compatibility study was carried out. This FVIII coagulation factor is however not marketed in all European countries.
Only licensed polypropylene injection sets should be used, because treatment failure can occur as a consequence of human von Willebrand factor adsorption to the internal surface of some injection equipment.
6.3 Shelf life
3 years.
Chemical and physical in-use stability has been demonstrated for 24 hours at 25°C.
From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user.
6.4 Special precautions for storage
Do not store above 25°C. Store in the original package in order to protect from light. Do not freeze.
6.5 Nature and contents of container
1 pack contains: powder in a vial (Type I glass) + 20 ml solvent in an injection vial (Type I glass) with a transfer system.
6.6 Special precautions for disposal
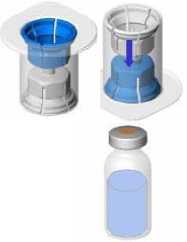
Reconstitution:
The currently applicable guidelines for aseptic procedures must be followed. The transfer system is only used to reconstitute the drug, as described below. It is not intended in administering the drug to the patient.
\

t

• Bring the two vials (powder and solvent) to ambient temperature (not above 25°C).
• Remove the protective cap from the solvent vial (water for injections) and from the powder vial.
• Disinfect the surface of each stopper.
• Remove the cap from the Mix2Vial device. Without removing the device from its packaging, attach the blue end of the Mix2Vial to the stopper of the solvent vial.
• Remove and discard the packaging. Take care not to touch the newly-exposed part of the device.
• Turn the solvent vial-device assembly over and attach to the powder vial using the transparent part of the device.
The solvent will automatically transfer to the powder vial. Hold the assembly and gently swirl to completely dissolve the product.

The powder generally dissolves instantaneously and should have dissolved in less than 10 minutes.
The solution should be clear or slightly opalescent, colourless or slightly yellowish. Cloudy solutions or solutions with deposits must not be used.
Administration:

• Hold the vial of reconstituted product in vertical position while screwing a sterile syringe onto the Mix2Vial device. Then slowly draw the product up into the syringe.
• Once the product has been transferred to the syringe, firmly hold the syringe (with the piston pointing downward), unscrew the Mix2Vial device and replace it with an intravenous or butterfly needle.
• Expel the air from the syringe and insert into the vein after disinfecting the surface.
• Inject slowly by intravenous route immediately after reconstitution as a single dose at a maximum rate of 4 ml/minute.
Any unused product or waste material should be disposed of in accordance with local requirements.
7 MARKETING AUTHORISATION HOLDER
LFB-BIOMEDICAMENTS 3, avenue des Tropiques BP 305 - LES ULIS 91958 Courtabreuf Cedex FRANCE
8 MARKETING AUTHORISATION NUMBER(S)
PL 28315/0007
9 DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
18/12/2014
10 DATE OF REVISION OF THE TEXT
18/12/2014